CrystalKitX 入門
CrystalKitX は、結晶構造の欠陥と界面構造をモデル化するための汎用プログラムです。あらかじめ定義した 1つまたは 2つの完全結晶または単位格子 (Crystal A とCrystal B で表します) を用いてモデルを作成します。 CrystalKitX は、MacTempasX のファイルおよびその出力ファイルを取り扱うことができます。
- インストールについて
- CrystalKitXの特徴
- メニューコマンド
- CrystalKitXのウィンドウ
- CrystalKitXのツール
- 基本原子と表示される原子の違い
- 事例:インジウムリンの双晶境界を生成
インストールについて
インストーラをダブルクリックすると、CrystalKitX とその関連ファイルをインストールすることができます。その際、管理者権限をもったユーザーアカウントを使用して下さい。CrystalKitX だけではなく、ハードウェアキーのドライバもインストールされます。
CrystalKitX は、ハードウェアキーを用いてライセンス管理を行っています。MacTempasX との共用キーを持っている場合は、CrystalKitX 用のハードウェアキーを USB スロットに差す必要はありません。そうでない場合は、キーボードまたはディスプレイ本体に付いているスロットに CrystalKitX のキーを差して下さい。
CrystalKitX をインストールし、最初に起動するとお名前、所属、ハードウェアキー番号を入力するダイアログが表示されます。使用するコンピュータを変更したり、OS をアップデートした場合は、ハードウェアキーのドライバを再インストールする必要があります。CrystalKitX のインストーラを再度実行して下さい。ドライバがインストールされていない場合は、ハードウェアキーの認識が行えず、したがって CrystalKitX はデモモードでしか動きません。
CrystalKitX の特徴
CrystalKitX は、結晶構造の欠陥と界面構造をモデル化するための汎用プログラムです。あらかじめ定義した 1つまたは 2つの完全結晶または単位格子 (Crystal A とCrystal B で表します) を用いてモデルを作成します。
CrystalKitX は、MacTempasX のファイルおよびその出力ファイルを取り扱うことができます。
CrystalKitX は、Crystal A と B (異なる単位格子でも、同一の格子でも可) を用いて、凝結結晶や界面/粒界を作成します。点欠陥、空洞、格子間原子などを描く場合は、どちらか1つだけで可能です。
CrystalKitX は、結晶格子や原子結合子などで構造を可視化します。その角度や距離は簡単に測定でき、断面も設定できます。 描画した原子は1つずつでもグループでも移動させることができ、カット&ペーストも可能です。またマウスクリックを行って、新たに原子を追加することもできます。結晶の回析パターンを表示することも可能です。
メニューコマンド
通常の Macintosh アプリケーションと異なり、CrystalKitX のメニューコマンドには Open… コマンドがありません。CrystalKitX の File メニューは、作成済みの CrystalKitX のファイルを開くためではなく、作成した構造や画像を保存するためにあります。以下に、CrystalKitX のコマンドの機能を説明します。
File メニュー
Window to Pict…
カレントウィンドウを PICT ファイルに保存します。
Write U.Cell to File…
単位格子定義ツールを使って作成した単位格子のデータを、MacTempasX のシミュレーションで使用するためにファイルに書き出しします。Crystal A または Crystal B として設定したデータを保存します。これにより結晶構造を変更した場合でも、元のデータを再度読み込むことができます。
Slice U.Cell to File…
単位格子を、Z 値で指定したスライス面で保存します。スライス面数は n で指定します。
Recreate Interface…
単位格子定義ツールで作成した単位格子を保存する際、2つの結晶の方位関係やその他の設定なども保存されます。このコマンドを使用してファイルを開くと、Crystal A と B を設定・保存したときと同じ状態で表示されます。
Page Setup…
印刷のためのページ設定です。
Print…
ウィンドウあるいはその中の選択領域を印刷します。
Editメニュー
編集メニューには、2つの機能があります。まず、その他のアプリケーションとの間でデータをやりとりするための Cut や Copy などの機能です。CrystalKitX の画像をコピー&ペーストで、他のアプリケーションに持っていゆくことができます。
その際、Option キーを押しながら Cut、Copy、Paste、Clear メニューコマンドを実行すると、ディスプレイ上で表示されている原子のみの操作ができます。この特殊機能を利用する場合は、必ずメニューコマンドを使用して下さい。キーボードコマンド (Apple+C キーなど) では利用できません。
Undo
搭載されていません。
Show Clipboard
クリップボードの内容を表示します。
Create A/B Precipitate
凝固結晶 A または B を作成します。まず 2種類の結晶の方位関係、界面、軸を設定します。次に Display メニューの中の Draw Penetrating Lattices を選択して、格子の差し込みを実行します。選択ツールのどれかを使って選択領域を設定し、Create A (または B) Precipitate コマンドをクリックすると、選択範囲内が A (または B) 、選択範囲外が B (または A) で凝固結晶の作成が行えます。
Preferences…
CrystalKitX の各種設定が行えます。その中には電子顕微鏡で使用する値や、MacTempasX がシミュレーションを行うときの層厚などがあります。
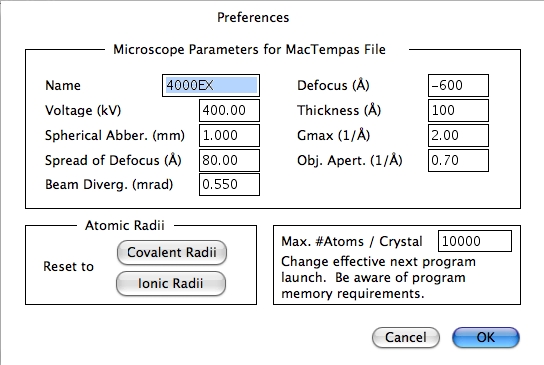
原子半径には、共有結合半径あるいはイオン結合半径を使うかデフォルト設定できます。個々の原子半径は、それぞれ変更することが可能です。取扱う最大原子数も設定できます。なお原子数を大きくとった場合、データサイズに対応したメモリ容量が必要となります。
Crystal Data メニュー
定義済みの Crystal A または B を読み込むためのメニューです。ファイルから読み込んだり、直接入力、あるいはすでに読み込み済みの構造を指定することができます。
Create Crystal A/B
ファイルのデータを読み込む場合は、MacOSX 標準のファイルダイアログが表示されます。Option キーを押しながら Read from File… コマンドを選択すると、EMS ファイルを読み込むことができます。ただしファイル名のチェックはなく、スーパーセルのみです。
通常の CrystalKitX ファイル (拡張子 [.at] ) は、MacTempasX と互換性があります。さらに、もし単位格子定義ツールによって作った結晶構造の場合は、ファイルのリソースフォークに2種類の単位格子のデータを保持しています。 データを手入力する場合は、以下のようなダイアログボックスが表示されます。
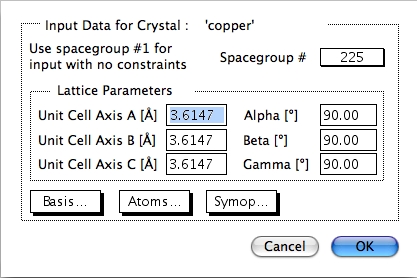
230 種類の空間群の中から結晶構造を指定する場合は、ポップアップメニューで結晶構造の種類を選択 (単斜晶、斜方晶、立方晶など) してから、 空間群を選択します。結晶の対象操作の回数は、CrystalKitX が自動的に行ってくれるので、格子定数を入力した後、原子を指定するだけで済みます。
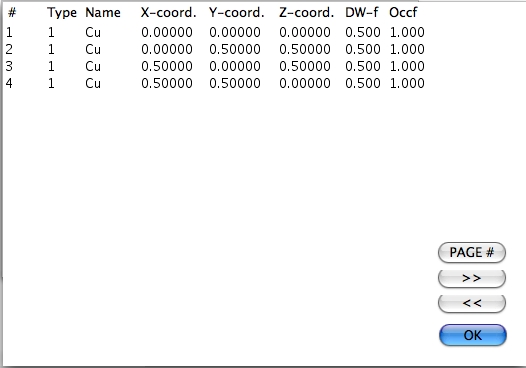
構造中の原子の総数と対象操作は、Atoms… および Symop… ボタンを押すと表示されます。
Show Crystal A/B
結晶構造のデータを表示します。結晶構造を入力するときと同じ様式で表示されます。データは修正することが可能です。
Orientation メニュー
このメニューには、1つのコマンドしかありません。
Set Orientation…
2つの結晶の方位関係を定義します。設定の仕方には 2通りあります。一般的に使用されるのは、2つの結晶の平行な面の方向で定義します。もちろん、それぞれ結晶の軸方向と回転角度を指定して定義することもできます。軸/角度のセットで定義するのは、それぞれの結晶が独立に設定されている場合に使用します。この場合は、特定の軸で回転する前に、2つの結晶は互いに平行な状態に置かれているものとします。
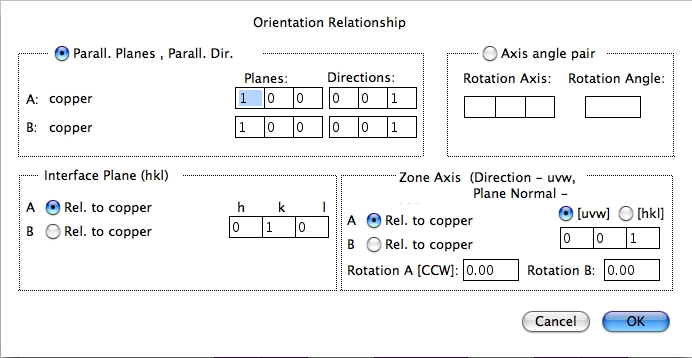
この方法では、双晶境界も簡単に設定できます。ダイアログは、方位関係の設定だけではなく、結晶と晶帯軸がなす界面を決めることもできます。この界面は 2つの結晶格子に対して、片側が Crystal A、反対側が Crystal B となる境界を表します。この界面は、Crystal A または Crystal B から求めることができます。晶帯軸を使って視野角を決めることができます。CrystalKitX では、現在のところ晶帯軸は界面に対して垂直でなければならいという制約があります。したがって、CrystalKitX で 傾斜界面を作成することはできません。
Display メニュー
結晶の断面を表示します。新しい単位格子を使って破断や格子間原子を描く場合、まず結晶を描く必要があります。2つの結晶の界面を描く場合は、界面を示す断面を作っておかなければなりません。
Draw Crystal A/B…
A または B どちらかの単位格子、またはどちらかの結晶の断面を表示します。
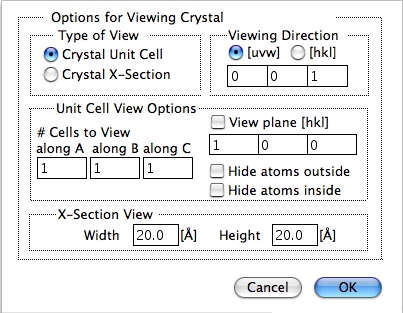
視野と晶帯軸は結晶断面とともに描かれ、1つだけ単位格子を表示している場合には晶帯軸はありません。1つの単位格子を描く場合は、つねに [001] の方位で描かれます。回転ツールを使って、任意の方向に回転させることもできます。
Draw X-Section…
設定した界面の断面を表示します。このコマンドは、2つの結晶の方位関係、界面、晶帯軸を設定した後でなければ使用できません。 晶帯軸を設定すると、視野角が決まります。
視野の設定値によって、描かれる結晶のサイズが決まります。界面は常に水平方向となります。界面が描かれると、2つの結晶はその面で接触できるようになります。界面には両方の結晶の原子が置かれます。 両方の原子が同じサイトの置かれ、他の原子によって見えなくなっている場合もありますが。なお Crystal A と Crystal B をどれだけ界面に近づけるか設定することによって、この重複を調節することができます。
Draw Penetr. Latt…(Draw Penetrating Lattices)
断面と同じ設定を使って格子の差し込みを描くことができますが、結晶は界面の両サイドに描かれます。これによって、2つの結晶がどのように重なり合うのか、わかります。このコマンドはまた、他の基質中での結晶の凝固を表すことができます。格子の差し込みを使って凝固結晶を描いたら、A または B の凝固結晶を作るために、Edit メニューを使用することができます。
Redraw Display
必要に応じて、ディスプレイを再描画します。
Make Movie…
Crystal A と B を 360 度回転させたムービーを作成します。ダイアログで Frame サイズを指定することができます。
Option メニュー
Interface Options…
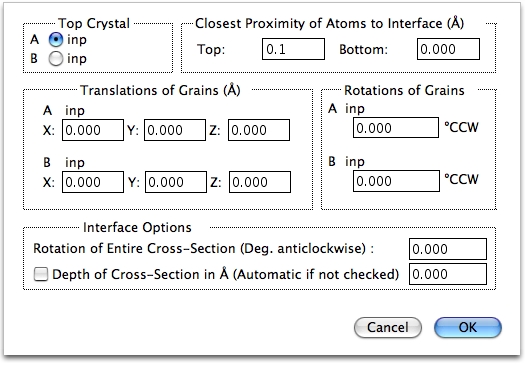
2つの結晶の界面を決定するための以下のパラメータが設定できます。
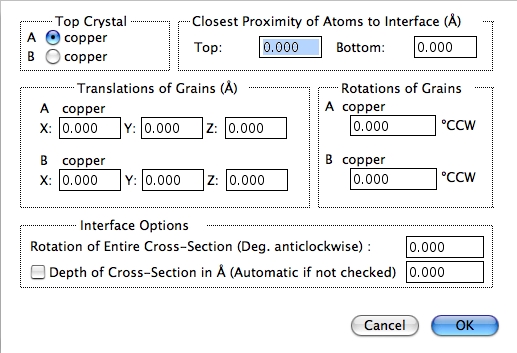
- Top Crystal
Crystal A と B のどちらが界面の上側になるか決定します。 - Closest Proximity of Atoms to the Interface
界面に対する原子の距離を設定します。両サイドの原子が界面に接触すると、2つの原子が同じサイトに位置した場合は、非現実的な構造となってしまいます。これは A と B に同じ結晶を指定した場合は、よく発生します。シミュレーション中に発生する不具合は、界面での過剰な原子に起因することよくあります。設定する単位はオングストロームです。 - Translation of Grains
結晶粒の変換コマンドは、界面を参照しながら 2つの結晶の変換を実行します。これらは剛体変換となります。単位はオングストロームです。 - Rotations of Grains
視野 (Z) 軸を参照しながら、A または B の回転を行います。回転は、方位設定ダイアログで設定された値に対してなされます。すなわち、結晶粒は最初設定された方位を向いており、次に設定された角度で回転をします。 - Rotation of Entire Cross-Section
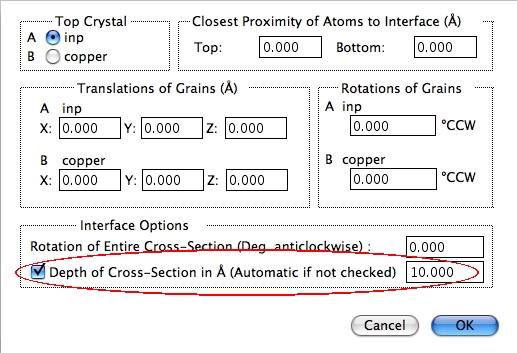
フィールド全体の視野をディスプレイの X 軸回りにわずかに回転させるオプションですが、めったに使用されません。X 軸は、界面の向きと同じで、ディスプレイの中の水平左側から右側に向かう軸です。 - Depth of Cross-Section
(1または 2つの結晶の) 断面を拡張するためのオプションです。通常、深度は晶帯軸の長さすなわち視野の Z 軸方向の長さに自動的に設定されます。しかしながら、チェックボックスを ON にし、オングストローム単位で数値を設定すると、変更することができます。正確にいえば、このオプションは単結晶の断面を表示するためのものですので、ここにあるべきではないかもしれません。深度は界面には関係ありませんから。
3D Atom Shading
Lighting Tool によって設定された条件で、原子を陰影付きの球体として表示します。
Atomic Bonds…
原子間の結合を、最大 5種類までユーザーが定義することができます。
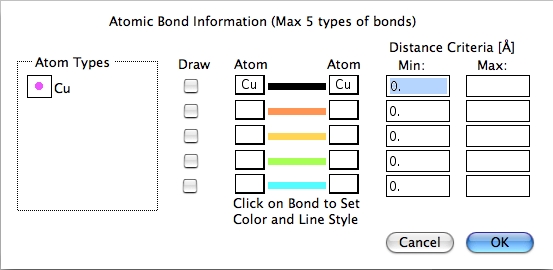
結合を描く 2つの原子を選択し、最小と最大間隔を設定します。 結合のラインをクリックすると、その色、スタイル、幅が設定できます。
Boundary Toleranse…
原子の相対座標が、どれだけ 0 または 1 に近いとき、境界 (0 と 1) にあるものとみなすか、設定します。通常は、0.001 に設定されています。

Remove Close Atoms…
2つの原子が接近しているとき、それらの原子を取り除きます。その接近距離を設定します。
Show Guides
グリッド線の表示を ON/OFF します。デフォルトのグリッド線は水平線と垂直線で、ポインタツールを使って自由に移動させることができます。ウィンドウ端のグレーの部分でマウスをクリック&ドラッグすると、グリッド線を追加することができます。界面を作成するときに、グリッド線があると大変便利です。
Real Lattice
ポップアップメニューが表示されます。デフォルト設定は Real Lattice で、Reciprocal Lattice またはそれら両方を選択することができます。
Recip. Lattice Option (Reciprocal Lattice)
Reciprocal Lattice オプションは、逆格子を表示するオプションです。パターンのスケールは、カメラによって決定されます。発散角度が、構造のスケールで決まる回析スポットサイズを変更します。
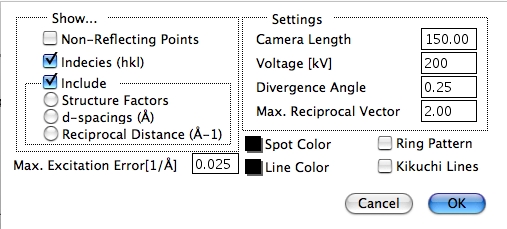
最大逆格子ベクトルは、Recip. Lattice Options… 設定ダイアログで設定できます。設定オプションには 1) パターンの指数 (hkl)、2) 構造因子、3) d-間隔、4) 逆格子ベクトルの長さがあります。Non-Reflecting Points オプションが ON になっていると、逆格子の中の無反射の位置を表示します。
Constrain Clicks to Atoms
単位格子の位置を選択したり、距離を測定する場合、通常はマウスを使用しますが、原子の内側をクリックしなければなりません。このオプションを OFF にしておくと、原子の位置に関係なくマウスを使用できます。
Draw Bonds out of Layer
断面あるいは界面が表示されているとき、ウィンドウ左下のポップアップメニューを使ってどのレイヤーを表示するか選択することができます。Draw Bonds out of Layer オプションが OFF のときは、表示されているレイヤー内の原子結合のみ表示されます。ON になっている場合は、表示されていないレイヤーの原子結合も表示されます。
Outline Atoms not in Layer
デフォルト設定では、表示されていないレイヤーの原子は非表示です。このオプションを ON にすると、他のレイヤーの原子も輪郭だけが表示されます。
Misc. (Miscellaneous) メニュー
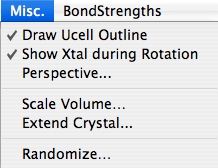
Draw UCell Outline (Draw Unit Cell)
結晶軸を描くかどうか決定します。デフォルト設定では、結晶軸とセルの境界が描画されます。
Show Xtal during Rotation (Show Crystal)
デフォルト設定では、回転ツールを使用した際、リアルタイムにその様子を描画します。このオプションを OFF にした場合は、結晶を回転させマウスを放したときにその様子が描き変えられます。
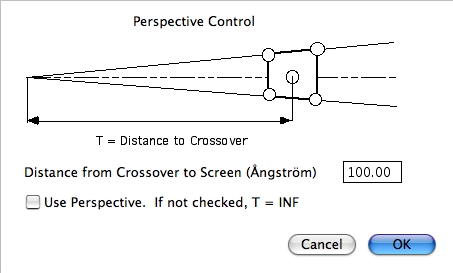
Perspective…
クリックすると Perspective Control ダイアログが表示されます。Use Perspective オプションが ON になっていると、スクリーンから焦点までの距離をユーザー指定の値に設定することができます。
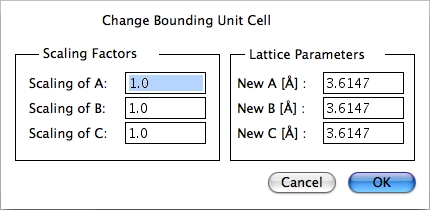
Scale Volume…
クリックすると Change Bounding Unit Cell ダイアログが表示されます。New A、B、C に数値を入力することで、単位格子のサイズを変更することができます。
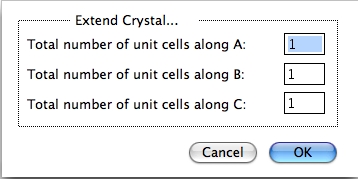
Extend Crystal…
先ほど変更した単位格子の指数を変更することができます。
Randomize…
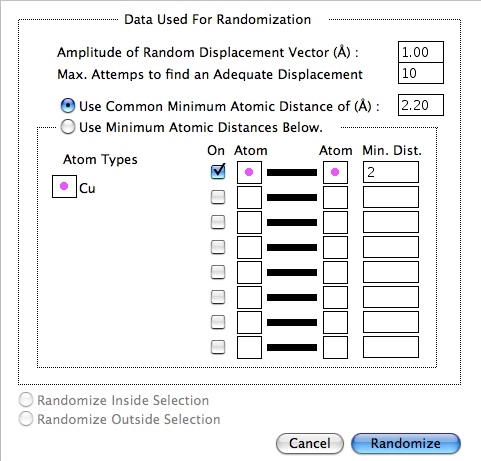
上記のダイアログを使って、各方向の原子をランダムに変更してアモルファス構造を作成するのは、あまりよい方法ではありません。デフォルト設定では、方向毎にランダムな値で原子が配置され、配置できる原子の最大数で決まる原子間距離よりも小さな値をとることはありません。もし原子間距離が原子の最大数と合わないときは、自動的に前回の値を使用し、該当する原子の数を表示してくれます。また、特定の原子間の最小距離を設定することもできます。
Atom Typesのセクションで原子を選択し、Atom のボックスをクリックして原子を設定し、距離を指定します。特定のサイズのアモルファス構造を作成するには、まず断面を作成し、単位格子の境界を決めた後、ランダム化します。Options|Constrain Click to Atoms オプションを OFF にする必要がありますが、ランダム化した後も単位格子をマークしておくことができます。
CrystalKitX のウィンドウ
CrystalKitX は、4つのウィンドウがあります。原子 (Atom) ウィンドウ、情報 (Info) ウィンドウ、ツール (Tools) ウィンドウ、メインウィンドウです。
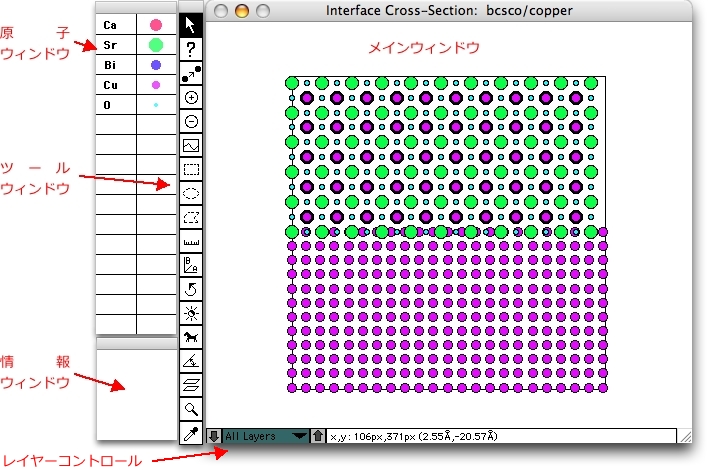
原子ウィンドウ
結晶に含まれている原子の種類を表します。相対原子半径は、原子の種類毎に異なる色が付けられている円の半径で表されます。円をダブルクリックすると、原子半径の値を変更することができます。 色選択ツールがアクティブなとき円をクリックすれば、ポップアップウィンドウの中から好きな色を選ぶことができます。
情報ウィンドウ
原子間の距離や角度など、結晶面の情報を表示します。メインウィンドウ下部のステータスラインに、補足方法が表示されます。
ツールウィンドウ
CrystalKitX の動作をコントロールするための各種ツールが用意されています。
メインウィンドウ
単位格子などを表示するためのウィンドウです。結晶断面は、ウィンドウ下部にあるレイヤーコントロール (上下の矢印に挟まれています) によって、特定のレイヤーを選択することができます。
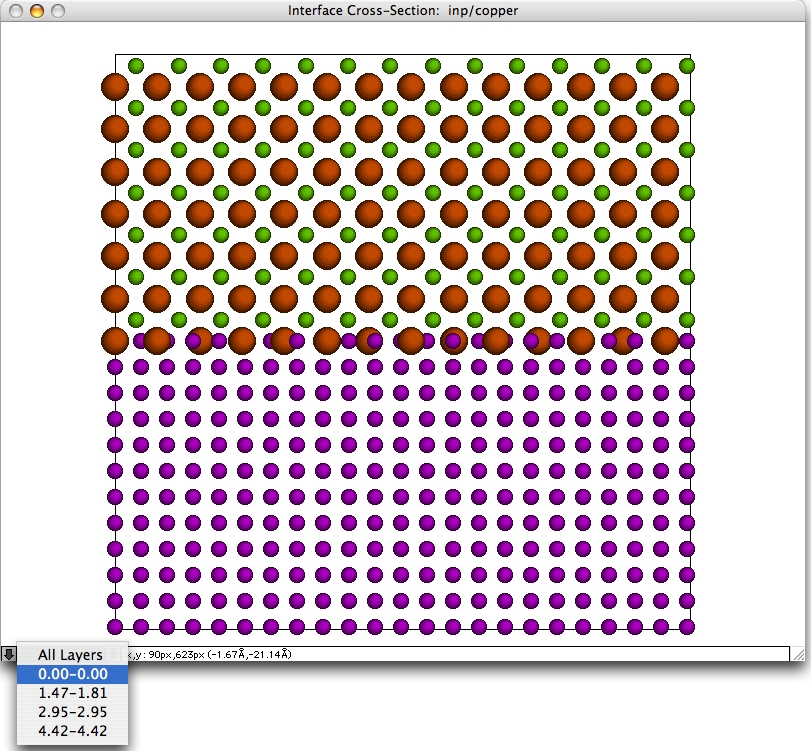
コントロールはポップアップメニューになっており、原子のレイヤーをオングストロームで表示します。レイヤーの中のどれかを選択すると、そのレイヤーに含まれる原子だけ表示されます。その原子を編集した場合、選択したレイヤーに含まれる原子のみが変更されます。
CrystalKitX のツール
CrystalKitX で利用できるツールは以下の通りです。該当する個所をクリックすると使用できるようになります。他のツールを選択するまで使用でき、カーソルの種類で区別することができます。

ポインタ
他のツールがアクティブでないとき、ポインタツールが利用できるようになります。特別なツールが何も選ばれていないことを表す以外、特に機能はありません。グリッド線を引いたり移動するときに、ポインタツールを使用します。
情報ツール
特定の原子上でこのツールをクリックすると、その原子に関する情報が表示されます。通常はデータ編集可能なダイアログが表示されます。 界面が描かれている場合は、情報ウィンドウが表示されます。Option キーを押してクリックした場合は、通常のダイアログが表示されます。
原子の移動ツール
界面または断面が描かれている場合のみ、原子を移動することができます。これらのツールで気をつけていただきたいのは、再描画 (Display|Redraw Display) すると変更個所が元に戻ってしまうことです。
原子の追加ツール
マウスクリックをした位置に原子を追加します。ツールをアクティブにすると、原子の種類を選択するダイアログが表示されます。
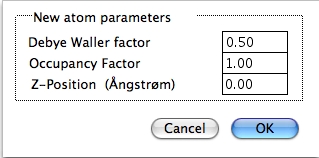
さらに、Debye-Waller 因子、占有因子、原子の z 方向 (視野の垂直方向) 高さを設定するダイアログが表示されます。
原子の削除ツール
個々の原子を削除しますが、1つの単位格子 (Crystal A または B) だけ表示している場合は、削除できません。
界面ツール
界面を含む断面図を描く際、任意の形状の界面をこのツールで指定できます。結晶のプロットの左側領域で始点をマークし、右側に界面のポイントをマークしていって、最後に右側領域で終わります。マークは左側に後戻りすることはできません。終点をマークすると、設定したポイントに沿って界面が作成されます。
選択ツール
凝固結晶をカット、コピーするために、任意の形状の領域を設定します。Shift キーを押しながら選択すると、矩形または円が描けます。領域を定義するためには、始点と終点が一致しなければなりません。
ルーラーツール
原子間の距離を測定するために使用します。通常、原子のある位置でクリックする必要があります。原子が見つからない場合は、警告音が鳴ります。Option キーを押しながらクリックした場合は、原子の位置に制約されません。
単位格子定義ツール
どちらかの結晶あるいは界面の断面図が作成されると、構造ファイルに書き出すための単位格子を設定することができます。まず原点を指定し、次に単位格子のベクトル (A)、つぎにベクトル (B) を指定します。Write U.Cell To File コマンドを使って、この単位格子を構造ファイルとして書き出しできます。単位格子を連続して設定した場合は、後の格子データで前の格子データが置換えられます。構造の変更、たとえば原子を移動したり削除した場合は、ファイルにもその変更が反映されます。断面を描き変えた場合は、格子データは失われます。通常、カーソルは原子の上でしか使用できませんが、Option キーを押しながらクリックすると、原子以外でのサイトでも利用できます。

※ 注意:晶帯軸方向の繰り返し距離は、Crystals A および B と同じではない場合があります。繰り返し距離の最大値は C 軸方向となり、他の結晶の繰り返し距離のみ原子が埋められます。短い構造での原子の占有因子は、より大きな C 軸を補正をするために変更されます。もし短い方の結晶で、他方の結晶の繰り返し距離まで原子を埋めることができるようになったら、結晶は正しくない数だけの単位格子をもち、回析パターンを計算するとき間違った反射が表れてしまうでしょう。C 軸は、Interface Options… メニューの Depth of Cross-Section の値を使って任意の値に固定することができます。
回転ツール
結晶の単位格子が表示されているとき (Crystal A/B)、好きな方向に単位格子を回転させることが できます。メインウィンドウでマウスボタンをクリックし、そのままマウスをドラッグします。Shift キーを押しながら動かすと、スクリーンの垂直方向を軸に回転します。
照明ツール
3D オプション付きで原子を描画しているとき、光源を設定します。メインウィンドウでマウスボタンをクリックし、押したまま移動させます。カーソルを追いた位置から少し離れたところに、球の陰を表す円が表示されます。
アニメーションツール
結晶の単位格子が表示されているとき (Crystal A/B)、単位格子を回転表示させます。メインウィンドウでマウスをクリックすると回転が始まり、再度クリックすると終了します。単位格子の描画に時間がかかるようなら、長時間回転させない方がよいでしょう。
角度ツール
2方向 (3原子) の角度を計測します。
面ツール
2つの原子をクリックして、面を指定します。
拡大鏡
1回クリックする度に 10% ずつ単位格子が拡大されます。縮小する場合は Option キーを押しながらクリックします。Shift キーを押しながらクリックすると 100% ずつ変更されます。
色選択ツール
原子の塗り色を変更します。原子ウィンドウの色付きの円をクリックすると、16 種類の色の中から好きな色を選択できるようになります。
基本原子と表示される原子の違い
Crystal A および B の構造は、ユーザーが変更し、Save Crystal A/B コマンドを使って保存することができます。通常、基本原子 (basis-atoms) リストといっしょに保存されます。このリストを表示するには、Show Crystal A/B コマンドをクリックします。
基本原子リストのみ変更が可能です。これは、原子リストが基本原子リストに対象操作を加えて作成されるからです。対象演算子が x,y,z (一致演算子) だけならば、原子リストと基本原子リストは同一です。ただし、プログラムは 2つのリストを有します。
1つの単位格子の構造を表示させると、ウィンドウ隅の原子リストにも原子が表示されます。たとえば基本原子リストに原子 が1つだけある (x=0,y=0,z=0) とき、これを編集して (1,0,0), (1,1,0), (1,1,1), (0,1,0) などの原子を作ったとき、スクリーンにはそれらの原子も表示されます。これらの構造は、Save Crystal A/B コマンドを使って保存することができます。
これとは異なる方法で構造を編集することもできます。1つの単位格子をスクリーン上に作成したら、回転ツールを使って格子を回転させます。情報ツールを使って原子をクリックすると、その情報が得ることができます。このとき、その値を変更したり原子の種類を変えることができます。変更した情報は原子リストに保持されます (基本原子リストではありません) 。これを Save Crystal A/B コマンドを使って保存しても、変更はファイルに保存されません。しかしながら、Option キーを押しながら保存すると、この原子リストも保存され、変更がファイルに保存されます。
なお、(1,1,1) の原子は変更できないことに注意して下さい。(1,1,1) の原子はどちらのリストにも保持されず、ただ単位格子の隅を表示するために使用されます。また、隅にある原子で単位格子内に存在するのはその 1/8 だけです。(0,0,0) の原子のみ全体が単位格子内にあります。その他の隅にある原子は、7/8 は隣の単位格子内に存在することになります。
事例:インジウムリンの双晶境界を生成
CrystalKitX を起動し、Crystal Data|Create Crystal A|Read From File… メニューコマンドをクリックし、InP.at ファイルを開きます。続いて Crystal Data|Create Crystal B|Same As A コマンドをクリックします。2つの結晶の定義が終わったので、方位関係を設定します。Set Orientations|Orient. Relations… メニューコマンドをクリックし、以下のように設定します。
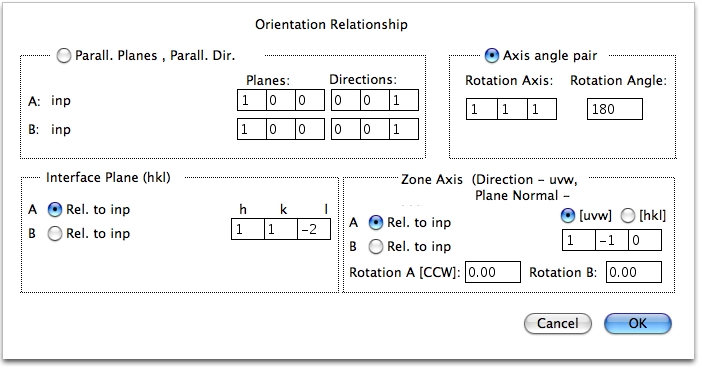
ダイアログの入力が終わったら OK ボタンを押し、Display|Draw XSection… メニューコマンドをクリックして、視野の範囲を 30×40 (オングストローム) に設定します。
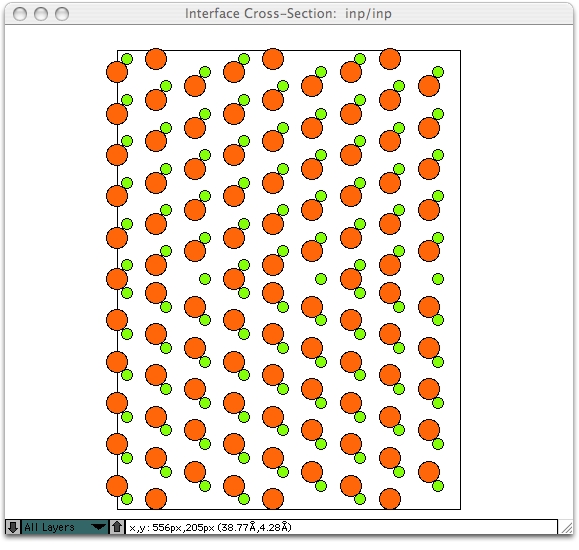
以下のように表示されるはずです。
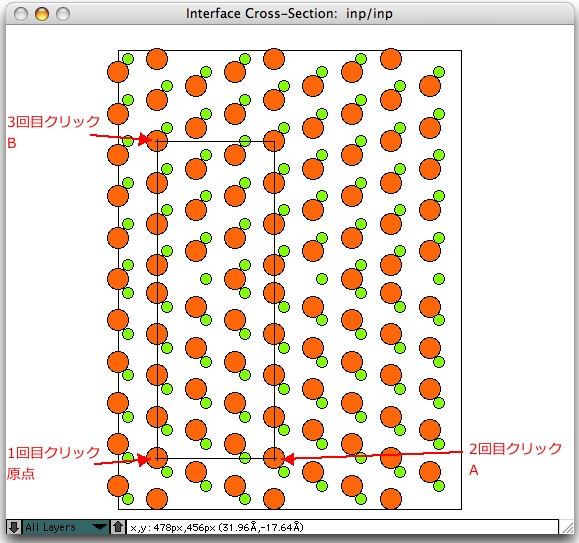
単位格子を設定するため、単位格子設定ツールを選択して原点と A、B 点を設定します。この時点では、両方の結晶が界面で接触するのが許されているため、界面上に 2セットの原子が表示されます。
これらの原子を削除するために、Options|Interface Options…のClosest Proximity of Atoms to Interface の Top を 0.1 に設定します。ウィンドウが書き替えられ、1セットの原子が界面上に表示されます。