日本語
English
株式会社ヒューリンクス
TEL:03-5642-8384
営業時間:9:00-17:30
分子動力学法
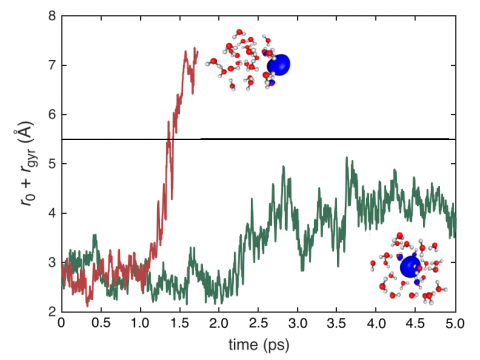
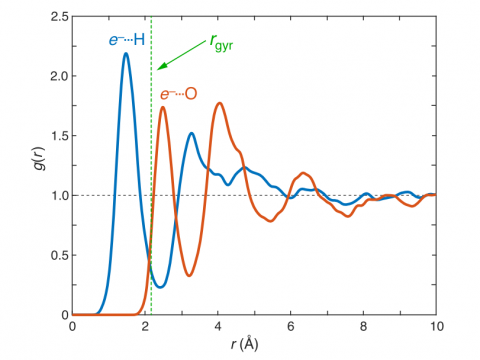
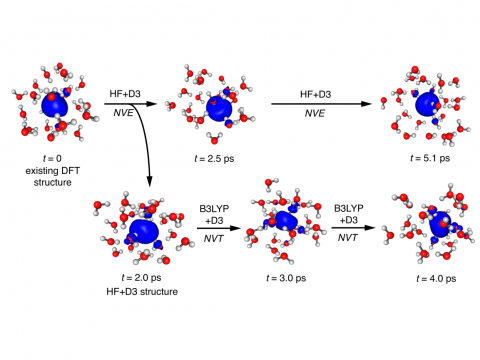
Q-Chem の第一原理分子動力学 (AIMD) や準古典的分子動力学 (QMD) は、研究課題が純粋な量子力学的アプローチでは対応できない少しばかりダイナミックなものである場合や、酵素のモデリングプロジェクトで何かエキサイティングな新しい軌道が用意されているような場合に、興味をそそるシミュレーションかもしれません。大規模な系に対して、Q-Chem には QM/MM や埋め込み機能が組み込まれており、CHARMM や Amber のような分子動力学シミュレーションソフトウエアパッケージとのインターフェースも備えています。反応経路の発見、 タンパク質とリガンドの結合エネルギーの予測、活性部位への基質の結合のモデリングを行うことができます。
Ab Initio 分子動力学法 (AIMD)
- NVE アンサンブル (デフォルト)
- NVT アンサンブル
- Langevin サーモスタット
- Nosé-Hoover サーモスタット
- 振動スペクトル
- 準古典的分子動力学 (QMD)
- 最少スイッチ表面ホッピング (FSSH)
- Ab Initio 経路積分法