日本語
English
株式会社ヒューリンクス
TEL:03-5642-8385
営業時間:9:00-17:30
M-Chem: 生物学的システムのモデリング
M-Chem は、大規模な生体分子システムのモデリングのための最新鋭のパッケージです。
主な特徴:
- 分子動力学 (MD) シミュレーションのための高性能 MPI/OpenMP ハイブリッド実装
- 固定電荷(Amber)、分極(AMOEBA)、および ReaxFF 力場
- 能勢=フーバーサーモスタットとバロスタット、共役勾配自己無撞着場、および、多体力、周期境界条件、Particle Mesh Ewald を解くための新しい拡張ラグランジュスキーム
- 標準的な力場やタンパク質-水シミュレーションにおいて、パラメータ割り当てと系の設計を簡単かつ再現性の高い方法で効率化するフロントエンドモジュール
- ReaxFF との QM/MM 統合
- 他のコードで解析が可能な軌跡のフォーマット
M-Chem に含まれる機能
AMOEBA 力場
AMOEBA 力場は分極効果を組み込むことで、分極を考慮しない従来の力場よりも高い精度を実現しています。
Python インターフェース
M-Chemには、入力処理および系の溶媒和のための、Python ベースのインターフェース機能が組み込まれています。
ReaxFF
ReaxFF は、結合の切断や形成を正確にシミュレーションできる反応性力場であり、反応や材料のモデリングに有用です。
QForce
QM 計算のデータを用いて、汎用的な力場を拡張するための、自動化されたオープンソースのツールキットです。これにより、計算コストを増やすことなく、力場の精度を向上させることが可能になります。GitHub リポジトリはこちらからご覧ください
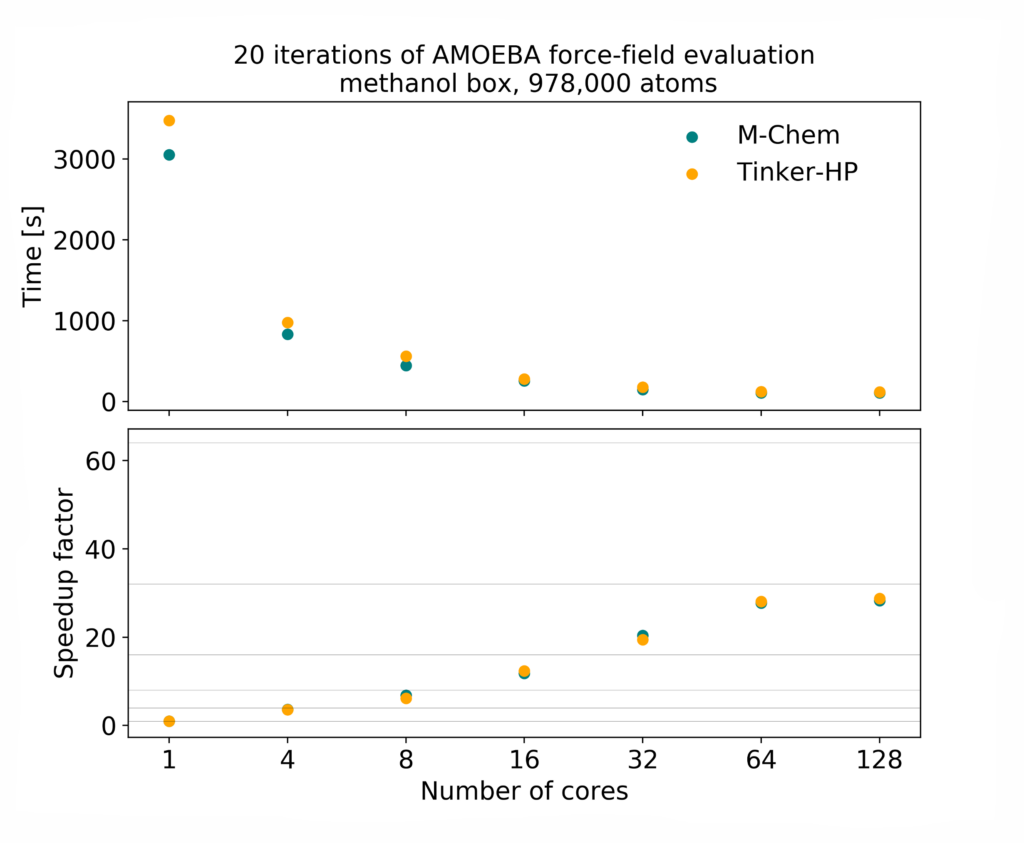
高速な並列処理性能
- ハイブリッド MPI/OpenMP 機能
- AMOEBA 力場を用いた分子動力学
- 能勢=フーバーサーモスタットとバロスタット
OpenMP 機能
- AMOEBA および MBUCB 力場を用いた分子動力学
- AMOEBA を用いた一点計算(例:モンテカルロシミュレーション)
- AMOEBA を用いた真空分子動力学シミュレーション
- 共役勾配法および拡張ラグランジュ法(iEL/SCF および iEL/0-SCF)を用いた AMOEBA モデルの誘起静電項の評価