日本語
English
株式会社ヒューリンクス
TEL:03-5642-8384
営業時間:9:00-17:30
Constrained DFT
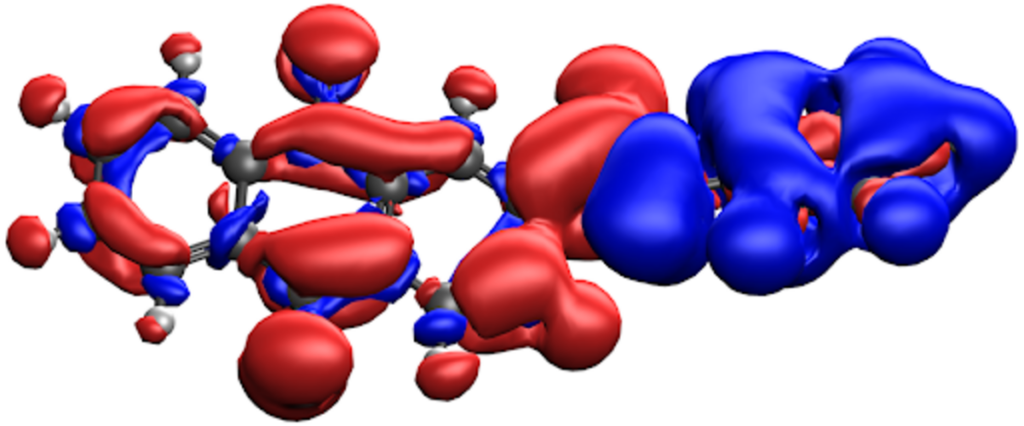
CDFT は、DFT 計算において Kohn-Sham ハミルトニアンにポテンシャルを追加し、電荷局在状態を得ることで、電子移動反応における断熱状態の近似に利用することができます。
- 標準的な SCF 計算ではアクセスできないような非断熱状態を構築し、対応する電子結合や他の電子移動パラメータを計算するための強力なツールです。
- Q-Chem では、異なる分子フラグメントに対して、全電荷制約とスピン電荷制約の 2 種類の制約を提供します。
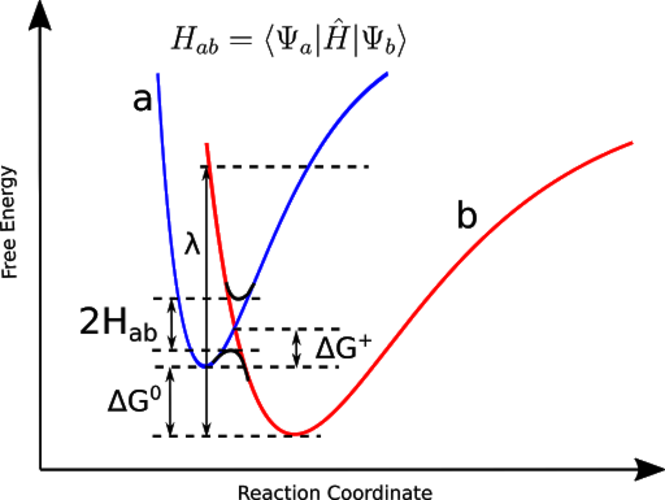
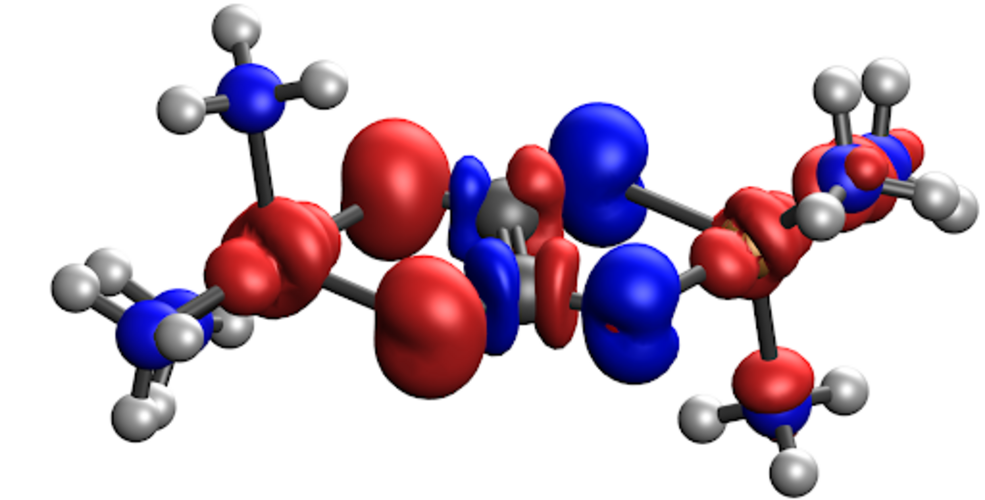
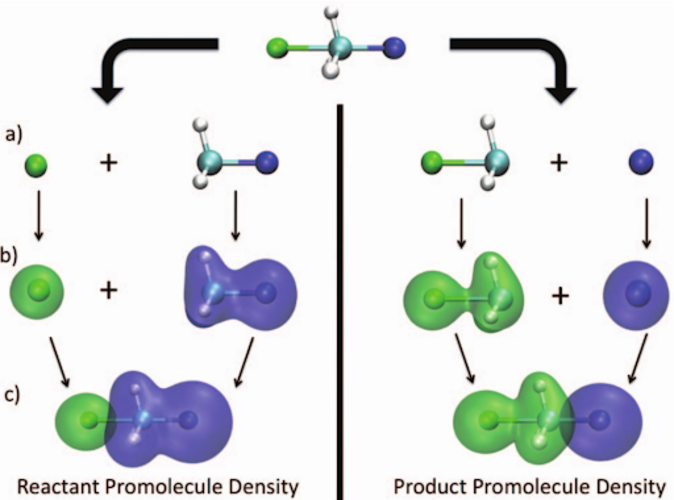
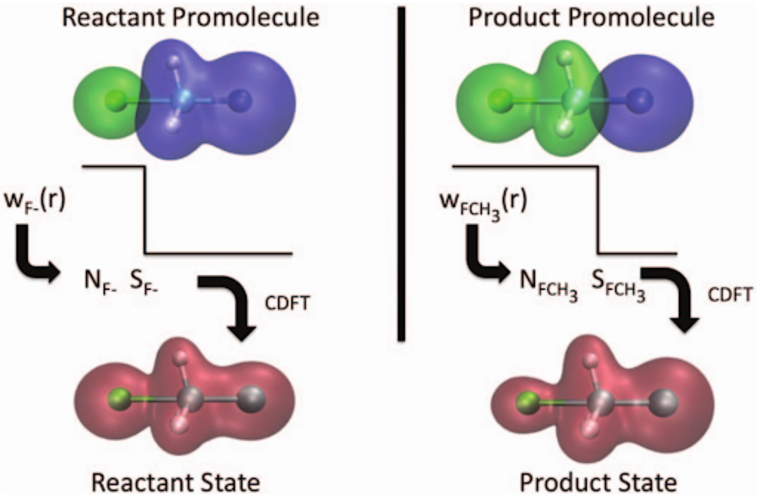
CDFT-CI による反応障壁の高さの予測
- 遷移状態のエネルギーを、反応物と生成物の 2 つの非断熱配位に架かる配位空間において探索します。
- 反応物および生成物の配位は、DFT 計算において電荷密度およびスピン密度の制約を適用し、反応物および生成物の電子的特性を最大限に保持することによって得られます。
- CDFT-CI は、従来の DFT と比較して反応障壁高さの計算値を大幅に向上させます。