Gaussian 03 製品概要
※この情報は旧バージョンの参考資料として掲載しているものです。必ずしも最新バージョンの仕様と一致しないことを予めご了承ください。
計算化学による研究領域を拡張
Gaussian 03 は、電子構造プログラム Gaussian シリーズの最新バージョンです。Gaussian 03 は、化学者、化学工学技術者、生化学者、物理学者をはじめ化学に携わる様々な分野の研究者に利用されています。
Gaussian は、量子力学の基本法則から、エネルギー、分子構造、分子系の振動数を予測します。また、これらの基本的な計算の種類から導かれる様々な分子特性も予測できます。安定種はもとより、短寿命中間体や遷移構造のような実験的に観測が困難であったり不可能であったりする化合物まで、広範な状態の下における分子の研究やその反応の研究に利用できます。
メインウィンドウ
大型分子組織の反応とスペクトルの研究
従来、タンパク質などの大型の生体分子は、電子構造法の対象外とされてきました。しかし、Gaussian 03 の ONIOM 法は、そのような制限を払拭します。Gaussian 98 より実装された ONIOM は、Gaussian 03 でいくつかの重要な改良が施され、より大型の分子に適用できるようになりました。
この計算方法は、分子の構造中に設定した2~3の層を異なるレベルの精度で処理することによって、巨大な分子をモデリングします。この予測された結果は、高精度の計算法によって生成されるものと本質的に同等であることが、検定調査によって証明されています。
Gaussian 03 の ONIOM 計算は、2次連立のアルゴリズムと micro-iterations 使用により構造最適化のパフォーマンスが向上しています。
加えて、このプログラムのONIOM 計算内に電子埋め込みを含めるオプションは、分子全体の立体特性と静電気特性を可能にし、例えば、酵素の活性部位のような高精度の層におけるモデリングプロセスまで考慮に入れることができます。これらの技術は、実験結果と非常によく一致する分子構造および特性の結果を生み出します。
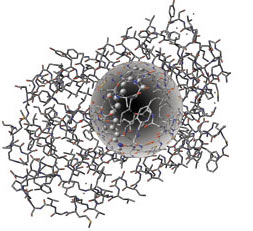
例えば、研究者は現在、この種が細胞内でどのようにしてエネルギーを生成するかを理解する最初のステップに ONIOM (MO:MM) モデルを使用して、最新のバクテリオロドプシン(図参照)の励起状態を研究しています。
この2層型アプローチにより、活性部位は電子構造法を利用して扱われ、残りの系は分子力学によってモデリングされます。電子埋め込み (活性部位の QM 法計算によるタンパク質環境の静電学を含む) は、分子の紫外-可視スペクトルの精密な予測に欠かすことができないものです。
ONIOM 法は、また、酵素反応、有機体系の反応メカニズム、表面のクラスタモデルおよび表面反応、有機体種の光化学プロセス、有機体および有機金属化合物の置換基効果および反応性、および同一触媒など、他の多くの領域の巨大な分子に適用可能です。
ONIOM に関するその他の新しい機能:
- 分子力学力場のカスタマイズが可能
- 効率的な ONIOM 振動計算
- 電気特性および電磁特性の ONIOM 計算
Spin-Spin 結合定数による構造の決定
構造解析は、X 線構造が使用できないような新たな化合物を研究する時において困難な問題が生じます。NMR スペクトルの磁気遮蔽データは、分子内における様々な原子間の結合に関する情報を提供します。Spin-Spin 結合定数は分子構造のねじれ角に依存するので分子の構造の特定に利用できます。
Gaussian 03 は、従来使用できた NMR 遮蔽および化学シフトに加えて、Spin-Spin 結合定数を予測できます。異なる構造を対象に定数を計算したり、予測されたスペクトルと実際に計測されたスペクトルを比較することで、計測された構造を特定することできます。
周期系の研究
Gaussian 03 では、PBC (周期境界条件) 法により高分子や結晶のような周期系としてモデリング可能な化学系の範囲が拡張されました。PBC 技術は、構造や化合物のバルク特性を確定するため反復するユニットセルとしてこれらの系をモデル化します。
Gaussian 03 は、例えば、高分子の平衡構造や遷移構造を予測することができます。また、異性化エネルギー、エネルギー反応過程などの予測による高分子の反応を研究することも可能で、分解や鉄の酸化も考慮に入れることができます。また、Gaussian 03 は、化合物の禁制帯のモデル化も可能です。
Gaussian 03 その他の PBC の能力
- 2D PBC 法は、界面や触媒作用の反応のような界面化学のモデルに利用できます。また、Gaussian 03 では、界面モデル・クラスタモデルを利用した同じ問題に、同じ基底系と Hartree-Fock 又は DFT 理論計算法を利用して研究することができます。Gaussian 03 を使えば、研究する系のための適切なアプローチが可能です。各モデルごとに能力や限界を考慮して枠組みを作り変える必要はありません。
- 3D PBC:結晶やその他の三次元の周期系の構造と可能なバルク特性を予測できます。
スペクトル予測
Gaussian 03 は、以下のような広範囲のスペクトルおよび分光特性を計算できます。
- IR/ラマン
- Pre-resonance ラマン
- 紫外-可視
- NMR
- 振動円偏光二色性 (VCD)
- 電子円偏光二色性 (ECD)
- 旋光分散 (ORD)
- 調和振動-回転結合
- 非調和振動と振動-回転結合
- gテンソルと他の超微細構造スペクトルのテンソル
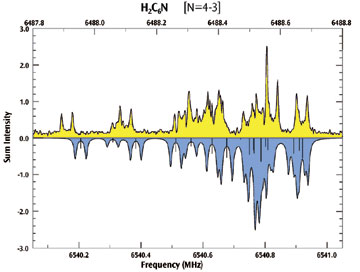
例えば、Gaussian 03 は、超微細構造スペクトルに有効なテンソルの多くを計算できます。これらの結果は、実験データからひとつを決定するのが困難な計測されたピークのスペクトル割り当てに有用です(以下の例を参照)。
反応と分子特性における溶媒効果のモデリング
分子特性と化学反応は、気相と溶液の間でその性質がしばしば相当に変化します。低構造の場合は、気相と溶液(および異なる溶媒)でかなり異なるエネルギーをもちえますし、構造平衡も異なり、反応もきわめて異なる経路をとります。
Gaussian 03 は、溶媒中の系のモデル化のために PCM (Polarizable Continuum Model) を提供します。このアプローチは、溶媒を分極連続体として表し、溶質を溶媒内の孔へ配置します。
Gaussian 03 の PCM 機能には、以下に示すような様々な機能強化が図られています:
- 溶媒中における励起状態の励起エネルギーと関連特性の計算
- NMR スペクトルとその他の磁性
- エネルギーの解析的2階導関数による振動周波数、IR およびラマンスペクトル、その他の特性の計算
- 分極率と超分極率
- パフォーマンスの全般的な向上
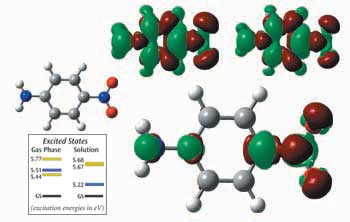
これらの表面は、パラニトロアニリンにおける基底状態と電荷移動励起状態の間で異なる電子密度を示します。右上の小さな表面は、気相における電子密度差を、その左側は、アセトニトリル溶媒における差を示します。電子密度は、励起状態において緑のエリアから赤のエリアに移動します。下の大きい方の表面の小さなものは、これら異なる密度の差(溶媒マイナス気相)です。溶媒中における基底状態から励起状態へのNH2からNO2への電荷移動が、気相におけるものに比べていかに大きいかを示します。また、グラフに示されるように、一番下の2つにオーダーされた励起状態は、気相からアセトニトリル溶液へ変化します。(黄色は 0 振動強度で、通常の紫外-可視スペクトルでは測定されません。)
Gaussian 03 で利用可能な計算手法
| SP, Scan | Opt, Force | Freq | IRC | ADMP | Polar | Stable | ONIOM | SCRF | PBC | |
| Mol. Mechanics | ● | ● | ● | ● | ||||||
| AM1, PM3 etc. | ● | ● | ● | ● | ● | |||||
| HF | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● |
| DFT methods | ● | ● | ● | ● | ● | ● | ● | ● | ● | ● |
| CASSCF | ● | ● | ● | ● | ● | ● | ● | |||
| MP2 | ● | ● | ● | ● | ● | ● | ||||
| MP3, MP4(SQD) | ● | ● | ● | ● | ||||||
| MP4(SDTQ), MP5 | ● | ● | ||||||||
| QCISD, CCD, CCSD(T) | ● | ● | ● | ● | ||||||
| BD | ● | ● | ||||||||
| OVGF | ● | |||||||||
| CBS, G*, W1 methods | ● | |||||||||
| CIS | ● | ● | ● | ● | ● | ● | ||||
| TD | ● | ● | ● | |||||||
| ZINDO | ● | ● | ||||||||
| SAC-CI | ● | ● | ● | |||||||
| CI | ● | ● | ● | ● | ||||||
| GVB | ● | ● | ● | ● |
Linda for Gaussian
Gaussian を簡単にクラスタ環境で実行
Gaussian と Linda を合わせて用いることによって、手軽に Gaussian の計算を複数のマシンに分散させることが可能です。
Linda for Gaussian による Gaussian のパラレルコンピューティングは、Gaussian 社と SCA 社 (Scientific Computing Associates, Inc.) との共同開発により実現したものです。Gaussian と Linda を合わせれば、通常のネットワーク上にある複数のコンピュータを容易に並列計算に組み込ませることができ、これにより非常に高いパフォーマンスが得られます。化学者は、これまであきらめていた興味深い様々な問題を研究することが可能になります。Linda for Gaussian は、下記の計算を並列処理できます:
- Hartree-Fock 法による構造最適化と振動数の計算
- 密度汎関数法による構造最適化と振動数の計算
- MP2 によるエネルギー計算と構造最適化
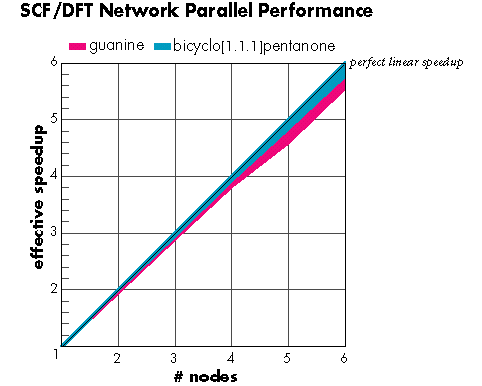
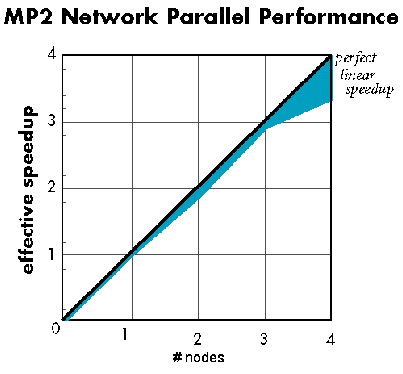
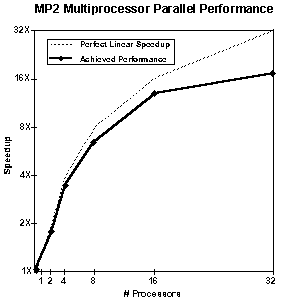
下図はイーサーネットで接続されている数台の Unix ワークステーションで得られる並列化の効率を示しています (Hartee-Fock 法、密度汎関数法)。グラフから、ノード数の増加に対してほぼ直線的にパフォーマンスが向上していることがお分かりいただけます。ピンク色の部分は guanine (175 basis functions) のエネルギー計算、水色の部分は bicyclopentanone (240 basis functions) を Hartee-Fock 計算した場合のスピードアップの様子をそれぞれ表しています。