
規準座標振動の解析
VIBRATZ は、原子価力定数や Urey-Bradley 力場定数を利用して、あらゆる分子や結晶の規準座標振動を計算するソフトウェアです。
※ 製品は英語アプリケーションです。
開発元:Shape Software
機能一覧
入力
- 結晶・高分子の空間群、または分子点群をリストから選択
- 結晶の単位胞パラメータ
- 原子の座標、対称型又は全ての原子
- 原子型(原子番号など)や距離、角度に関する力定数の詳細設定
- 計算は内部座標(internal coordinates)ではなく、デカルト座標によってなされます。力定数の数を限定したり、冗長式を与える必要はありません。
- 力定数の詳細設定では、構造間の移動が容易に可能です。また、デカルトマトリックスのフォーム内に記述することも可能ですので、任意のポテンシャルエネルギー関数を利用できます。
出力結果
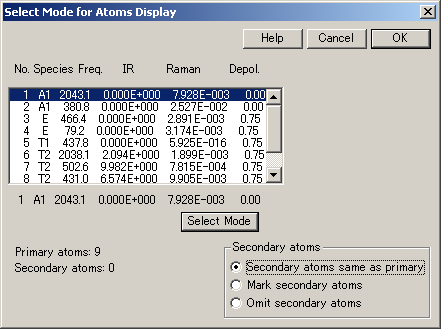
- 基本的な振動モードの振動数の全リスト
(結晶の場合は、optical or kappa=0 modes ) - 各モードにおける原子運動
- ポテンシャルエネルギーの分布
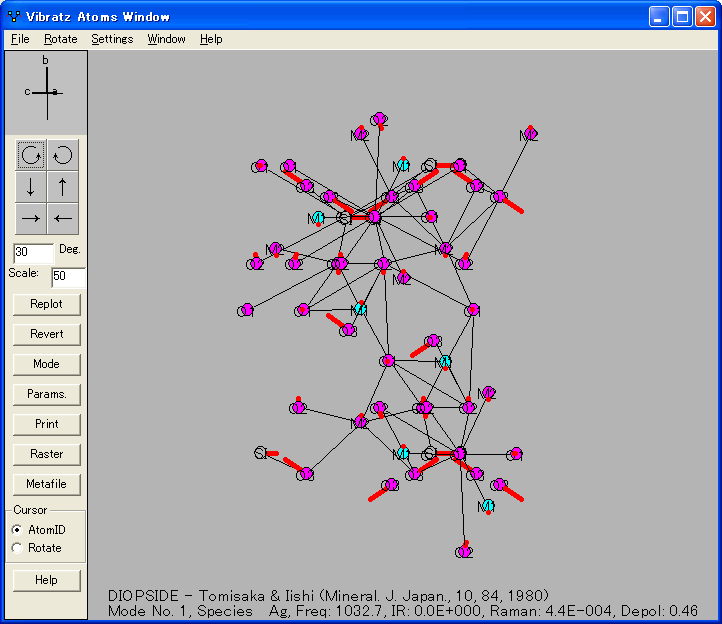
- 原子運動付き構造のプロット
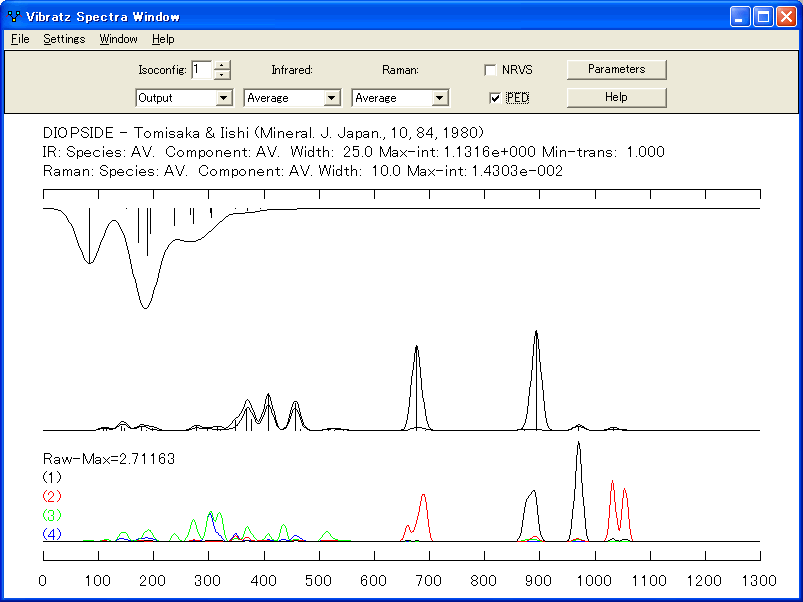
- 赤外線とラマンスペクトルの合成
VIBRATZ 製品概要
VIBRATZ は、化合物の原子価力 (原子間の結合と角度) を用いて光学的な基本振動をモデリングするソフトウェアです。VIBRATZ は基本振動数のみを計算し、オーバートーン(倍音)や、コンビネーション(結合)の計算は行いません(これらは、スペクトルにおいて通常は非常に弱い値をとるためです)。一般に、n 個の原子を有する分子においては、3n-6 個の基本振動が存在します。
- 結晶においては、光学的振動(又はより厳密に限定された光学的振動)は、その波数ベクトル(wavevector)が κ=0 として定義されます。このような振動においては、単位格子 (Unit Cell) はすべて等しく振舞います。これに対して、非光学的振動の場合は、異なる単位格子に存在する原子はそれぞれ独立した動きを示します。
- 光学的な振動モードの数は限られており、結晶の振動モードは 3n-3 個です。この場合、n は単位格子内にある原子の個数を示します。一方、非光学的モードの数は、実際の結晶に存在する全原子数に依存します。
VIBRATZ は、以下に示す5つを入力します。
1.結晶のための単位格子の軸長 (axis lengths) と軸角 (interaxial angles)
2.点群 (point-group)
又は空間群 (space-group) の対称性の選択又は入力。
3.原子座標 (atomic coordinates)
4.原子価力に関する結合 (Bonds) と角度 (Angles) の指定、および力の定数の指定
カテゴリ 4) における Bonds および Angles は、
伝統的な基準座標分析の内部座標(又はそれらのグループ)となります。
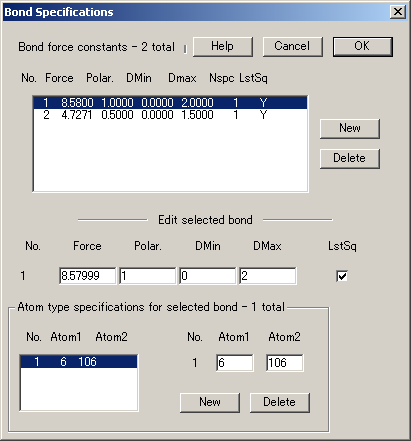
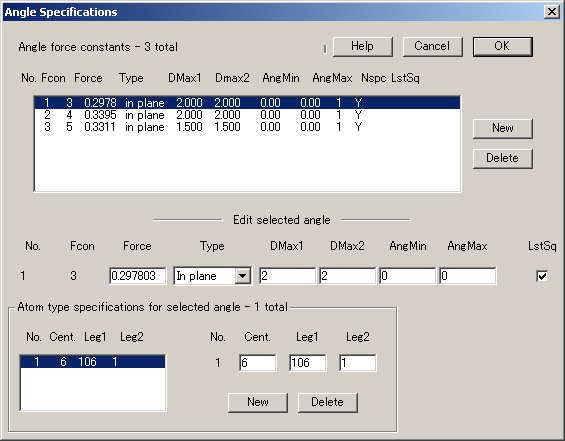
VIBRATZ は、全ての力を直交座標系(Cartesian)に変換し、直交行列を使用して対称性の解析を実行し、永年方程式 (secular equation) を解くため、そこで使用できる内部座標の数に制限はありません。また、内部座標間の重複性の関係を明示的に考慮する必要もありません。
結合および角度を設定するダイアログは、原子タイプおよび結合長と角度の範囲の指定によって成り立っています。原子タイプには、通常、化学種(原子番号)を入力します。ただし、結合と角度をより厳密に限定するために、それらは入力原子とは対称的に区別されたものになる場合もあります。Angles(角度の設定) には、4原子間のねじれ角(4-atom torsion)、結合面の曲げ(bond-plane bending)、3原子間の原子価角度(3-atom valence angles)が含まれます。内部座標の任意の対どうしの相互作用を指定できます。
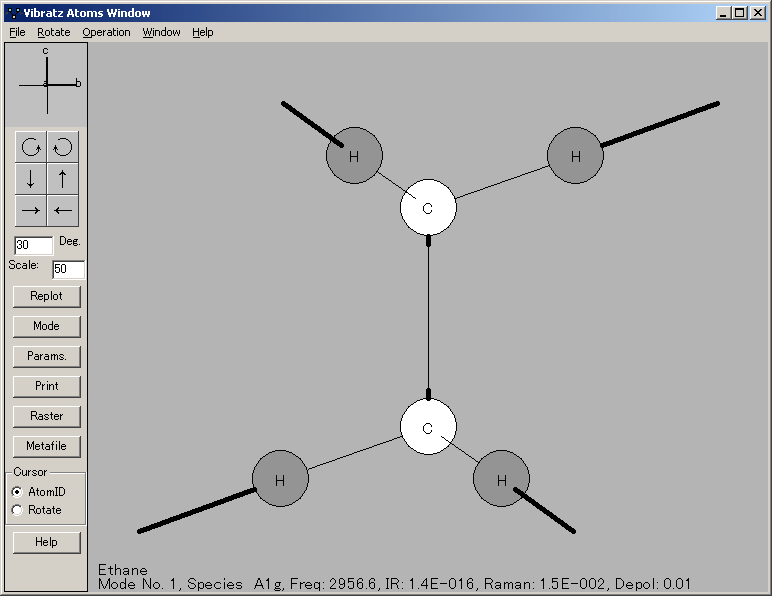
これらの情報が揃うと、VIBRATZ は、 対称性を分析し、振動数を計算し、対称種(symmetry species)を分類します。それは、各振動に含まれる原子変位と、結合や角度(およびそれらの相互作用)によって与えられる振動の全エネルギーの割合(fraction)を提供します。個々の原子の直交座標の変位という点からみると、原子間力の行列 (interatomic force matrix) は、この計算を通じて得られる副産物です。それは、弾性特性の計算等の別の目的のために利用されることもあります。
力の定数と対称種の両方の選択において、力の定数の最小二乗法による調整 (Least-squares adjustment) を実行できます。
シンプルなスペクトル強度モデルを使用して、赤外線スペクトルとラマンスペクトルの合成を描画することもできます。個々の偏光成分とスペクトルの平均を表示することもできます。原子の運動をグラフィカルに表示できます。
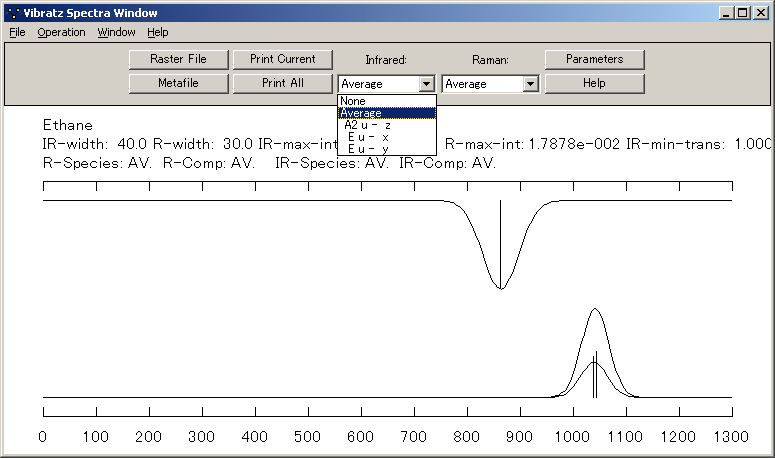
ラマンスペクトルの合成
その他の機能
- 原子価力場の複数定義と、Gaussian その他のプログラムで作成した Cartesian (直交) 力の原子価 (内部) 力への変換。EXAFS のための結合長の変動計算。
- ステップバイステップによる最小二乗法試行錯誤をともなう調節作業を、これにより事実上自動化することができます。力の定数は改善されなくなるまで一定量ずつ変更されます。これは実用面において、”Analytical (解析的な)” 最小二乗法よりも、非常に強力な手法を提供します。
- バッジャー則 (BADGER’S RULE)
Urey-Bradley や ligand-ligand の力の定数に含まれる FCON 内の特定の結合の力の定数やスペックを原子間距離の指数関数の形に変更します。
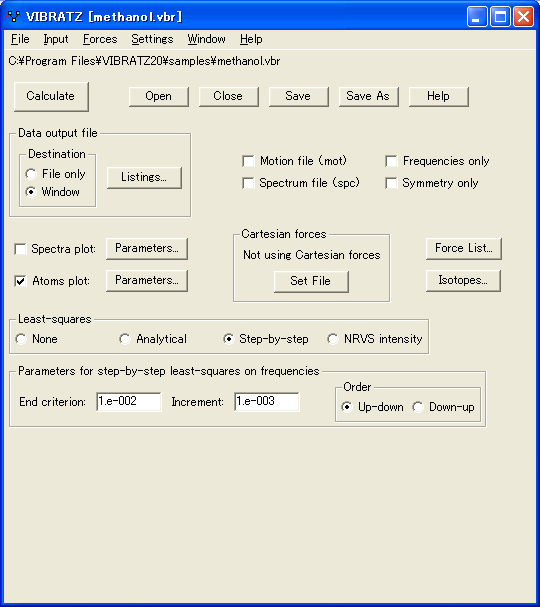
- (個々の原子の) 力の定数の手動設定
生成された原子の一つ一つの結合において、力の定数を指定できます。原子タイプや距離/角度の制限によるグローバルパラメータによって与えられる値に追加することも、置き換えることもできます。
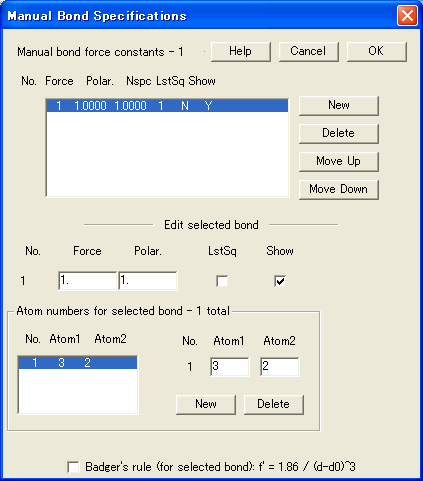
- PED (Potential Energy Distribution) 曲線
ポテンシャルエネルギー分布、単一または複数の力の定数による各モードのエネルギーに対する寄与を、スペクトルウィンドウに表示できます。
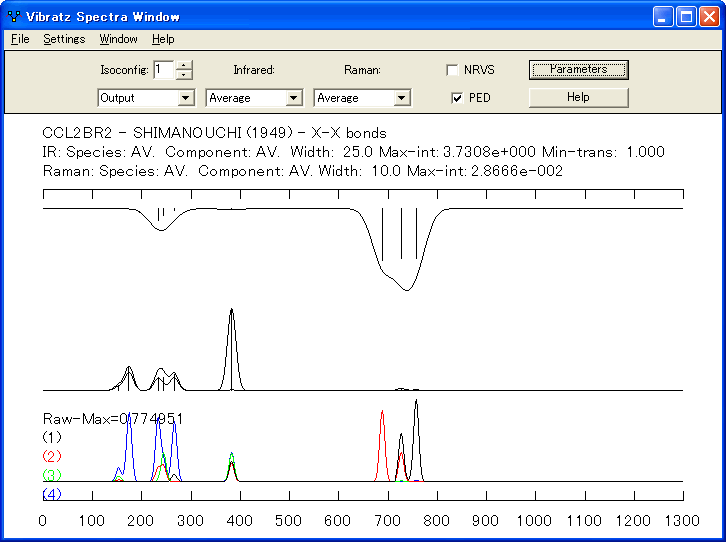
- NRVS (Nuclear Resonance Vibrational Spectroscopy) スペクトル
VIBRATZ は、ある原子のモード部分に基づく光学振動の状態密度を計算できるようになりました。これにより、核共鳴散乱の分子振動スペクトルのシミュレーションが可能になりました。通常それらは、Fe57 のガンマ線吸収により生成され、原子の振動に関する有効な情報を提供します。
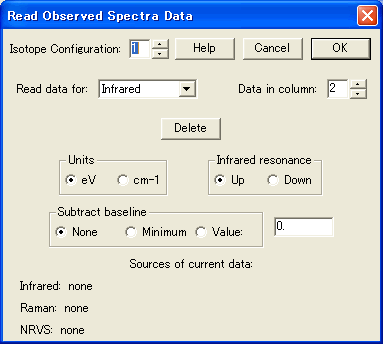
- 観測されたスペクトル
数値化された観測スペクトルをグラフィカルに読むことが可能となり、計算されたスペクトルと比較することができます。
- Z マトリックスによる入力
Z マトリックス形式で、分子の原子座標を入力できるようになりました。直交座標ではなく、結合距離と角度のシーケンスからなります。
- CIF ファイルのインポート機能の改良
CIF ファイルのインポート機能が改良されました。mmCIF (分子) フォーマットもサポートされました。
動作環境
VIBRATZ 2.3 動作環境
Windows
- Windows 10/11 (※ 製品自体 32 bit アプリケーションのため、64 bit 環境に最適化はされておりません。)
トライアル
デモ版お申込み
製品価格
製品価格
| 製品名 | 税込価格(円) |
|---|---|
VIBRATZ Win v2.3 | ¥ 29,700 |
お見積り・ご購入
※ アップグレードの場合
現在お持ちの製品名、バージョン、シリアル又はライセンス番号を必ずご記入ください。
価格につきましてはお問い合わせください。