Chem3D
分子モデリング/可視化アプリケーション
Chem3D は、分子モデリングと最先端のタンパク質可視化機能を有した化学者、および生物学者の両者に向けて開発されたアプリケーションです。タンパク質-リガンド複合体やDNA構造を3Dで詳細に可視化できます。また、水素結合やコノリー表面を表示し、これらを分析することも可能です。
ChemDrawパネルを用いると、パネル上で分子を描画すると同時に、これに対応する3D構造が即座に表示されます。分子の重ね合わせ (オーバーレイ) や、分子の立体配座の決定、MM2/MMFF94 による構造最適化など、基本的な分子モデリング計算を実行することもできます。
また、Chem3D Ultra に限りますが、MOPAC、GAMESS、Gaussian などのインターフェースを使用することで、ab initio計算や、半経験的計算、NMRの予測/可視化、IR/Ramanスペクトルの可視化なども実行が可能です。
| ChemDraw Prime | ChemDraw Professional | Signals ChemDraw | |
|---|---|---|---|
| 使用可能なアプリケーション (※ Windows版のみ) | – | Chem3D Pro | Chem3D Ultra |
| 化学的物性の予測 | ■ | ■ | |
| MM2 | ■ | ■ | |
| 立体配座空間の検討 (二面角) | ■ | ■ | |
| 非局在化電荷の表示 | ■ | ■ | |
| 原子のグループ化 | ■ | ■ | |
| 水素結合の表示 | ■ | ■ | |
| ケクレ構造と非局在化構造の切り替え | ■ | ■ | |
| スペクトル結果の表示 | ■ | ■ | |
| CONFLEX インターフェース | ■ | ||
| GAMESS インターフェース | ■ | ||
| Gaussian インターフェース | ■ | ||
| MOPAC インターフェース | ■ | ||
| AutoDock インターフェース | ■ | ||
| 分子モデルの高速オーバーレイ | ■ | ||
| MMFF94 | ■ | ||
| 部分的な溶媒接触表面/コノリー表面の生成 | ■ | ||
| 立体配座の決定 | ■ | ||
| Structure Browser | ■ |
Chem3D Pro 以上の共通機能
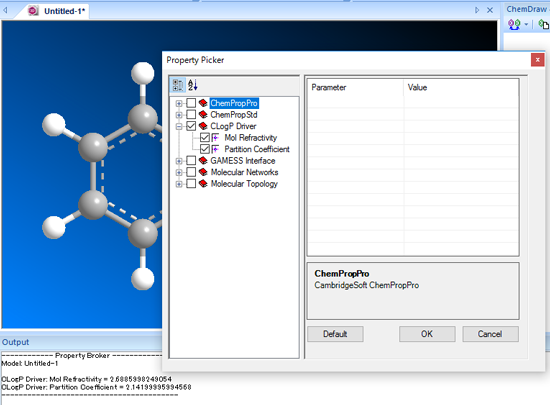
■ 化学的物性の予測
ClogP や、LogP、LogS、pKa といったさまざまな化学的物性値を予測することができます。
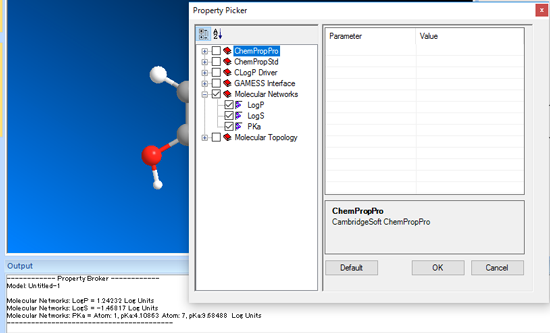
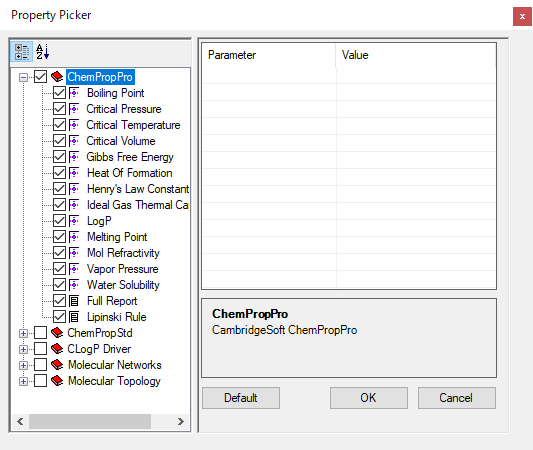
Chem3D Ultra では、より多くの化学的物性値予測が可能です。
■ MM2
Chem3D では MM2 をサポートしており、構造の最適化を行うことができます。
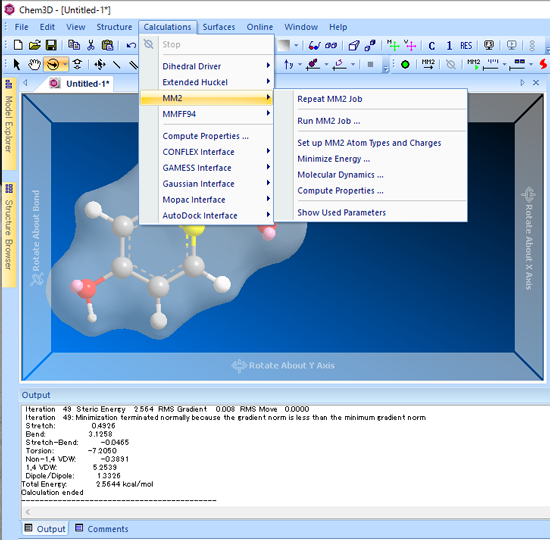
■ 立体配座空間の検討 (二面角)
二面角ドライバー (Dihedral Driver) を使用することで、最大で2つの二面角の角度を変えることができ、モデルの配座エネルギーを計算できます。
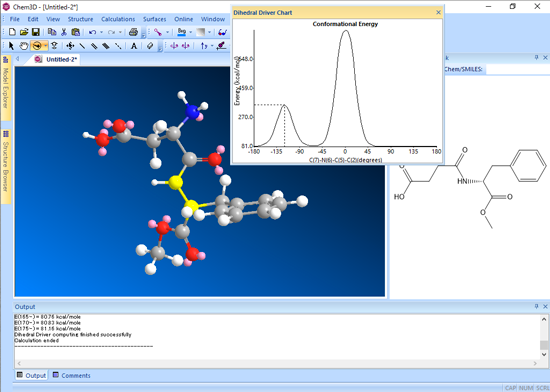
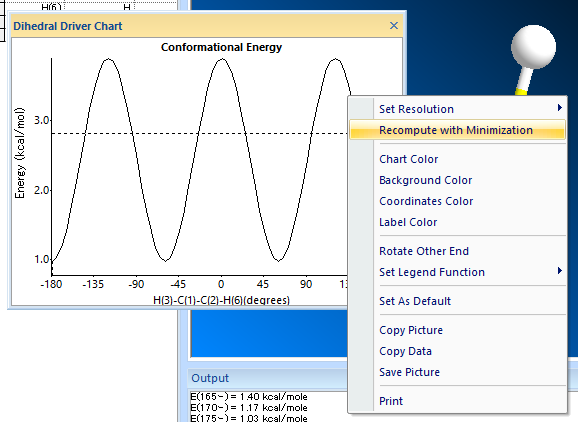
エネルギー値が最小の立体配座を特定し、これを開始点として更に微調整 (Recompute with minimization) を行い、安定した定常点を見つけることができます。
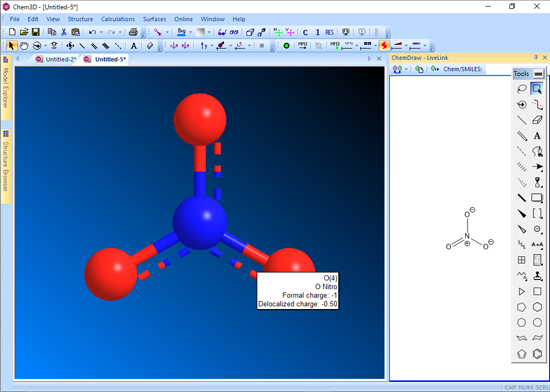
■ 非局在化電荷の表示
Chem3D では原子に形式電荷 (Formal Charges) を割り当てることによって、モデル内の非局在化が生じている全ての原子に対して、非局在化電荷 (Delocalized Charge) を計算します。
■ 原子のグループ化
モデル内の複数の原子を選択して、グループ化ができます。グループ化した原子団には、一括で色を付けたり、非表示にしたり、表示モードを変更したり、ラベルを設定することができます。
原子団を利用して、タンパク質の活性部位のような、モデルの特定部分を強調表示することも可能です。
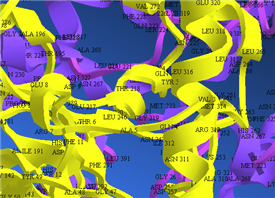
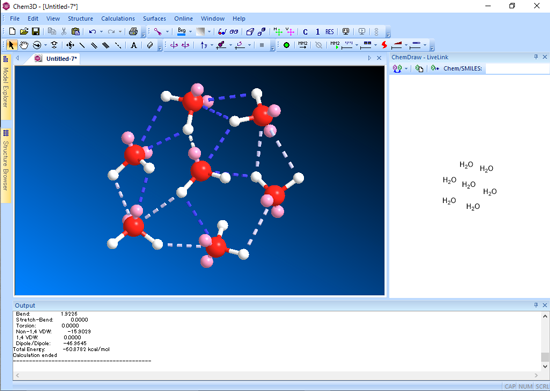
■ 水素結合の表示
Chem3D では、水素結合を表示することができます。
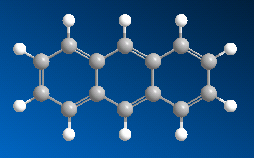
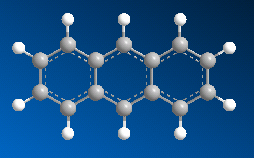
■ ケクレ構造と非局在化構造の切り替え
芳香族などの二重結合と単結合が交互に現れる化合物は、ケクレ構造、または非局在化構造として表示できます。Chem3D ではこれら構造を切り替えることが可能です。
■ スペクトル結果の表示
Gaussian、GAMESS 等で得られたスペクトル計算結果を、グラフとして別ウィンドウに表示します。
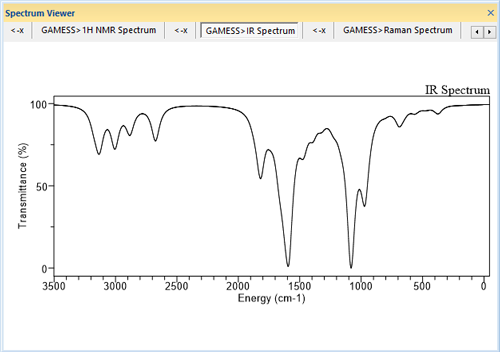
※ Gaussian、GAMESS 等は別売製品です。Chem3D には含まれていません。
Chem3D Ultra の機能
■ CONFLEX インターフェース
CONFLEX は、立体配座の解析パッケージです。CONFLEX インターフェースを使用することで、モデル内で安定した配座異性体を探索することができます。
CONFLEX インターフェースでは、以下の力場がサポートされています。
- MM2
- MM3
- EMM2
- MMFF
- MMFF94S
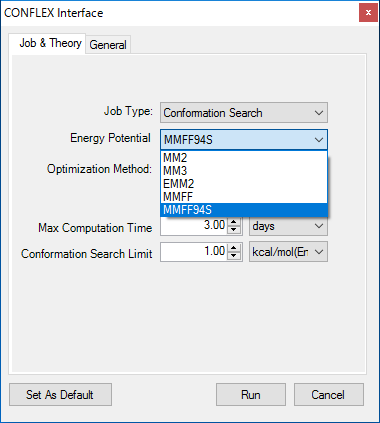
※ CONFLEX は別売製品です。Chem3D には含まれていません。
■ GAMESS インターフェース
GAMESS は、ab initio量子化学のパッケージです。アイオワ州立大学のGordon研究グループのメンバーによって維持管理されています。
GAMESS インターフェースを使用することで、RHF、ROHF、UHF、GVB、および MCSCF を使用するSCF波動関数の計算ができます。また、これらの中には、CIやMP2エネルギー補正計算で利用できるものもあります。
GAMESS インターフェースでは、以下の計算がサポートされています。
- エネルギーの最小化
- 遷移状態への最適化
- 物性特性の計算
- IR/Raman スペクトル予測
- NMR スペクトル予測
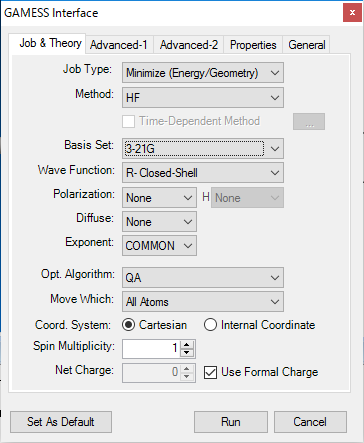
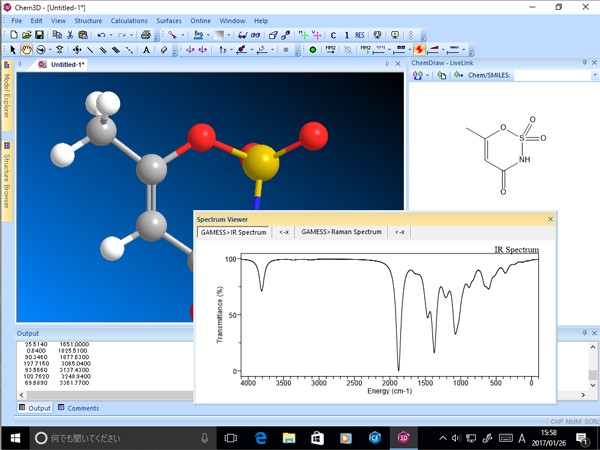
※ GAMESS は別途インストールする必要があります。Chem3D には含まれていません。
■ Gaussian インターフェース
Gaussian は、ab initio法と半経験的手法の両方を使用する計算化学のアプリケーションです。Gaussian インターフェースを使用することで、NMR、IR/Raman、および UV/Vis のスペクトルを予測できます。
Gaussian インターフェースでは、以下のような Gaussian におけるすべての計算がサポートされています。
- 1H/13C-NMR スペクトル予測
- IR/Raman スペクトル予測
- 複数のジョブステップ
- 部分構造最適化
- DFT 法のサポート
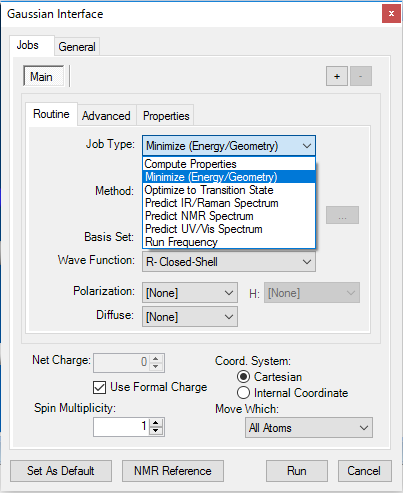
※ Gaussian は別売製品です。Chem3D には含まれていません。
■ MOPAC インターフェース
MOPAC は、半経験的手法に特化した計算化学のアプリケーションです。
MOPAC インターフェースでは、以下の機能がサポートされています。
- エネルギーの最小化
- 安定構造への最適化
- 遷移状態への最適化
- 物性特性の計算
- 双極子モーメント
- 電荷の安定性
- 分子内の電荷分布
- 分極率
- 気相/液相における安定性
- 超微細結合定数
- UHF スピン密度
- RHF スピン密度
- MOPAC ファイルの使用
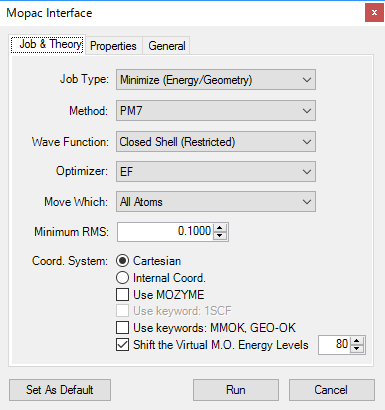
※ MOPAC は別途インストールする必要があります。Chem3D には含まれていません。
■ AutoDock インターフェース
AutoDock は自動ドッキングツールで、既知3D構造の受容体に対してどのようにリガンドが結合するのか予測できます。
AutoDock インターフェースを使用することで、受容体の準備、リガンドの準備、グループの定義、GPF の準備、DPF の準備等の複数のステップを経て、ドッキングの計算を実行することが可能です。各ステップは、AutoDock インターフェースのタブベースのダイアログにまとめられています。
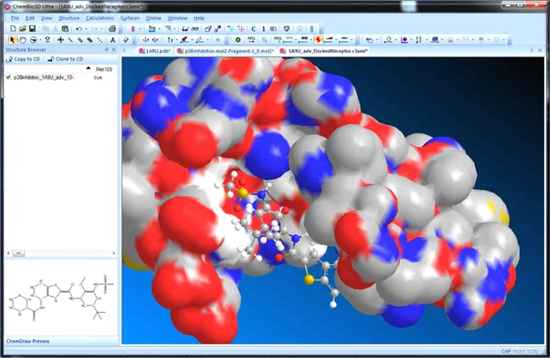
※ AutoDock はインストール支援メニューが用意されており、別途インストールする必要があります。Chem3D には含まれていません。
■ 分子モデルの高速オーバーレイ
Chem3D には2種類のオーバーレイ手法が用意されています。高速オーバーレイでは、2つの分子モデルから、いずれか一方をターゲット分子として指定することで、Chem3D が自動的に2つのモデルを重ね合わせます。
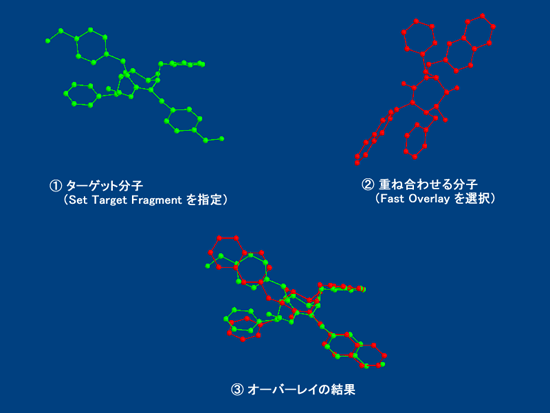
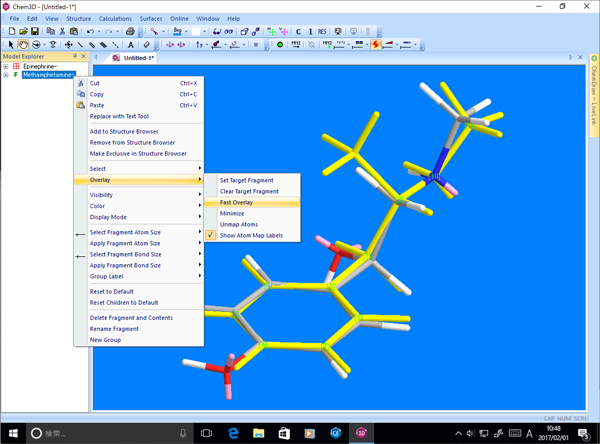
■ MMFF94
MMFF94 を使用して、分子モデルのポテンシャルエネルギーを計算できます。非結合エネルギーは、静電相互作用とファンデルワールス相互作用のエネルギーの総和になります。
- 静電相互作用の計算
静電エネルギーは、分子の非結合原子の電荷、それらの原子間距離、および分子の誘電率からなる関数で表され、空間的に近接した粒子や原子間の相互作用と、離れた位置にある原子間の相互作用が考慮されます。
Chem3D には、静電相互作用を近似する3つの方法 (Exact Method、高速多重極展開法 (Fast Multipole Method, FMM)、Adaptive Tree Code (ATC)) がサポートされているため、計算においてカットオフ法は一切不要です。
- ファンデルワールス相互作用の計算
ファンデルワールス相互作用の計算は、非結合エネルギーの計算で、非結合原子間の引力と斥力を考慮します。
原子数の増加に伴い計算にかかる時間は延びていきますが、Chem3D には、計算結果を近似する3つの方法 (シフト関数、スイッチング関数、トランケーション関数) が用意されています。
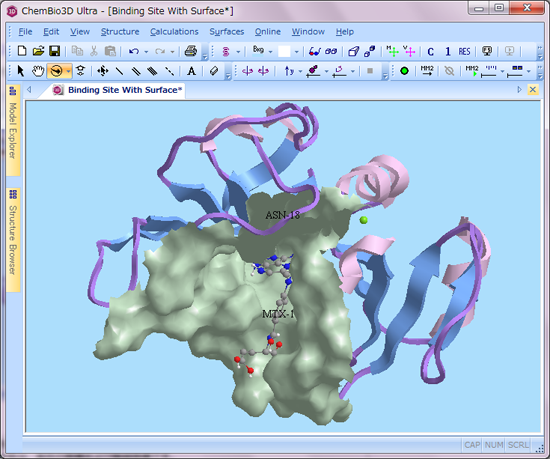
■ 部分的な溶媒接触表面/コノリー表面の生成
配位子を除いたタンパク質の分子表面や、タンパク質の活性部位を示す表面を作成することができます。作成した表面は、透明度や反射率、色等も設定することが可能です。
■ 立体配座の決定
Chem3D では、初期状態における構造や原子の座標、結合から分子の妥当な立体構造を決定することができます。
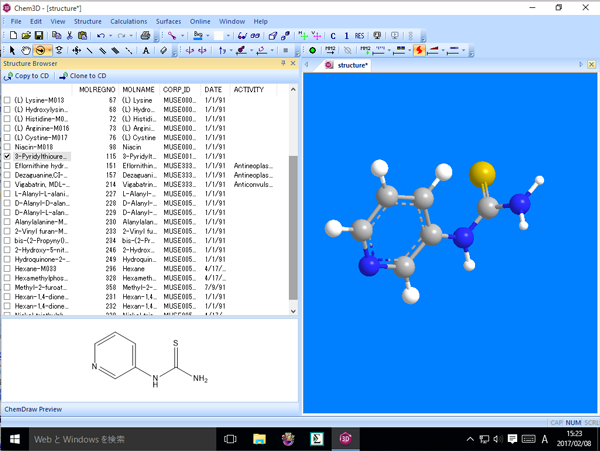
■ Structure Browser
Structure Browser は、ファイルに保存されている複数の構造式を参照し、それら構造式をモデルウィンドウに表示できます。また、表示する構造式を選択し、それらを重ね合わせたり、その性質を容易に比較することが可能です。