
汎用量子化学計算パッケージ
Gaussian 16 は、電子構造プログラム Gaussian シリーズの最新バージョンです。化学や生物化学、物理学をはじめとしてサイエンスの幅広い分野の研究者に向けて、電子状態計算や計算化学モデルについて最先端の手法や技術を提供します。利用可能なプラットフォームの種類は多岐にわたり、どのプラットフォームにおいても計算化学的な機能のすべてを使うことができます。さらに、GaussView 6 と合わせればより使いやすく、高度な機能を活用できます。
Windows 端末で動作する Gaussian 16 は Gaussian 16 W と呼ばれる製品です。Gaussian 16 W には、Linux/Unix バージョンの Gaussian 16 と同等にご利用可能な 64-bitバージョンの製品があります。また、32-bitバージョンでは、シュリンク ラップ・ライセンス (開封により使用許諾に同意したものとみなされるライセンス) として製品を購入することができます。macOS で使用する場合は、Linux/Unix バージョンの Gaussian 16 をお求めください。(Linux/Unix バージョンの Gaussian 16 の中に macOS で利用可能な製品が含まれています。)
Gaussian 16 では、マルチプロセッサ (マルチコア) 環境における共有メモリ並列 (ノード内並列) や、クラスタおよびネットワークを介した並列 (ノード間並列) 、さらにそれらを組み合わせた並列処理が可能です。ノード間並列処理には並列演算環境ソフトウェア Linda の最新バージョンのライセンスが別途必要です。
インストールや操作方法の支援サービス
- ヒューリンクスでは Gaussian/GaussView のインストールや基本操作などのサポートサービス (有償) をご提供しております。
詳しくはこちらをご覧ください。
最適な計算環境の構築の支援サービス
- ヒューリンクスでは、Gaussian/GaussView を実行するためのハードウェアも取り扱っております。
詳しくは弊社 (soft.sales@hulinks.co.jp) までお問い合わせください。
開発元:Gaussian, Inc.
機能一覧
モデル化学
手法と基底関数の組み合わせは、理論の水準を定め、Gaussian におけるモデル化学を決定します。すべての Gaussianジョブは手法と基底関数を指定しなければなりません。いくつかの手法では基底関数情報が指定済みですが、手法と基底関数は、通常、インプットファイルのルートセクションにおいて 2つの別々のキーワードによって指定されます。密度汎関数を用いたジョブでは、密度フィッティング (density fitting) 基底系を用いる場合もあります。密度フィッティング基底について詳しい情報は、Basis Sets をご覧ください。
下の表は Gaussian で利用可能な手法とジョブタイプの組合せを示しています。数値的にのみ計算可能なものを「num」で示し、解析的に計算可能なものをアスタリスク (*) で示しています。いくつかのキーワードについては表の下で議論します。
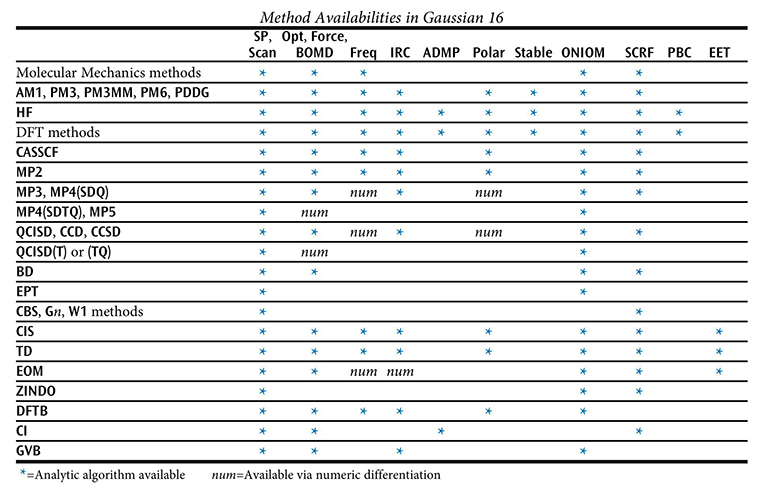
キーワードに手法が指定されない場合、HF とみなされます。多くの手法のキーワードは、例えば、ROHF、 UMP2 や RQCISD というように、手法の接頭語として、閉殻制限波動関数に R を、非制限開殻波動関数に U を、制限開殻波動関数に RO を置きます。RO は Hartree-Fock や密度汎関数法、AM1、PM3、PM3MM、PM6、PDDG によるエネルギーと勾配計算、MP2、MP3、MP4、CCSDエネルギー計算にのみ利用可能です。
通常、手法のキーワードはひとつだけ指定されるべきであり、2つ以上の手法を示すキーワードを含む場合には、奇妙な結果が生じるでしょう。しかしながら、以下は例外です。
- 動的な電子相関を含むCASSCF計算を要請するために、CASSCF は MP2 と一緒に指定できます。
- ONIOM および IRCMax ジョブは複数の手法の指定が必要です。しかし、それらは対応するキーワードのオプションとして与えます。
- model2//model1 という形式は、model1 での構造最適化計算に続いて、自動的に構造最適化された構造を用いて model2 での一点計算を行うためによく用いられます。
ジョブタイプ
以下は Gaussian 16 において利用可能なジョブタイプの一覧です。
- SP : 一点エネルギー
- Opt : 構造最適化
- Freq : 振動解析および熱化学解析
- IRC : 反応経路探索
- IRCMax : 特定の反応経路に沿ったエネルギー最大値の探索
- Scan : ポテンシャルエネルギースキャン
- Polar : 分極率および超分極率
- ADMP および BOMD : ダイレクトダイナミクストラジェクトリ計算
- EET : 励起エネルギー移動計算
- Force : 原子核上の力の計算
- Stable : 波動関数の信頼性テスト
- Volume : 分子体積の計算
- Density=Checkpoint Guess=Only : ポピュレーション解析のみを再計算
- Guess=Only : 分子軌道係数の初期値 (guess) のみを出力 (フラグメントベースの分子軌道係数の初期値の生成)
通常、ジョブタイプキーワードはただひとつのみ指定されますが、下記は例外です。
- Polar および Opt は Freq と組み合わせが可能です。後者の組み合わせの場合、構造最適化計算のあと続けて自動的に構造最適化後の構造を用いて振動数計算が行われます。
- 構造最適化計算へのオプション指定として、Opt は Compound法キーワードと組み合わせることが可能です。例えば、Opt=(TS,ReadFC) CBS-QB3 のように指定することが可能です。
ジョブタイプキーワードがルートセクションの中に指定されていない場合、デフォルトの計算タイプは通常、一点エネルギー計算 (SP) です。しかし、ルートセクションが method2/basis2 // method1/basis1 という形式であった場合には、 (method1/basis1レベルでの) 構造最適化計算に続いて、構造最適化後の構造を用いて (method2/basis2レベルでの) 一点エネルギー計算を要求するために使用されます。例えば、次のルートセクションは、B3LYP/6-31G(d) の構造最適化計算と、続けて CCSD/6-31G(d)モデル化学を用いた一点エネルギー計算を要求します。
# CCSD/6-31G(d)//B3LYP/6-31G(d)
このとき、Opt キーワードは、任意であり、デフォルトです。Opt Freq 計算はこの構文において用いることはできない点にご注意ください。
分子のプロパティ
以下に、要望の多い予測値とそれらを計算するための Gaussian 16 のキーワードを示します。
- 非調和の赤外 (IR) 吸収スペクトル、ラマン (Raman) スペクトル、振動円二色性 (VCD) スペクトル、ラマン光学活性 (ROA) スペクトル : Freq=Anharmonic
- 反強磁性のカップリング : Guess=Fragment、 Stable
- 原子の電荷 : Pop
- 溶媒和のΔGエネルギー : SCRF=SMD
- 双極子モーメント : Pop
- 電子親和力 : CBS-QB3、CCSD、EPT
- 電子密度 : cubegen
- 電子の円偏光二色性 : CIS、TD、EOM、SAC-CI
- 静電ポテンシャル : cubegen、Prop
- 電荷に由来する静電ポテンシャル : Pop=Chelp、Pop=ChelpG、Pop=MK
- 電子遷移バンド構造 : Freq=FranckCondon、Freq=HerzbergTeller
- 分極率、超分極率 : Freq、Polar、CPHF=RdFreq、Polar=DCSHG
- 高精度のエネルギー : CBS-QB3、G2、G3、G4、W1U、W1BD
- 超微細カップリング定数 (異方性) : Prop
- 超微細スペクトルテンソル (Gテンソルを含む) : Freq=(VCD, VibRot, Anharmonic)
- イオン化ポテンシャル : CBS-QB3、CCSD、EPT
- 赤外 (IR) 吸収スペクトルおよびラマン (Raman) スペクトル : Freq
- 前期共鳴ラマンスペクトル : Freq CPHF=RdFreq
- 共鳴ラマンスペクトル : Freq=ReadFCHT
- 分子軌道 : Pop=Regular
- 多極子モーメント : Pop
- NMR遮蔽定数およびケミカルシフト : NMR
- NMRスピン―スピンカップリング定数 : NMR=Mixed
- 旋光性 : Polar=OptRot
- ラマン光学活性 : Freq=ROA
- 熱化学解析 : Freq
- 紫外可視 (UV/Vis) スペクトル : CIS、ZIndo、TD、EOM、SAC-CI
- 振動回転カップリング : Freq=VibRot
- 振動円二色性 : Freq=VCD
- 振電スペクトル : Freq=ReadFCHT
プログラムの制限
ここでは Gaussian 16 に存在する様々なサイズの限界を示します。
- 積分のプログラムには以下の制限があります。
- 原子の最大数は 250,000個です。
- 原始シェルは全部で最大750,000個までです。
- Dまたはそれ以上の原始シェルは全部で最大250,000個までです。
- 縮約シェルの最大数は250,000個です。
- 縮約可能なシェルの数は最大で100個までです。
- Opt=(EF,EnOnly) 最適化は、解析的な勾配が求められない手法の場合に役立ちますが、変数の数は50個までに制限されています。
- GVB プログラムは 100対の軌道に制限されています (実際のところ、これは制約にはなりません)。
- Gaussian の内部の NBO 3.0 は、250,000原子かつ 10,000基底関数に制限されています。
リンク (Links)
以下は、リンク (links) として知られている Gaussian 16 を構成するプログラムとその基本的な機能です。
- L0: プログラムの初期化、オーバーレイの操作
- L1: ルートセクションの処理、実行するためのリンクの一覧の構築、スクラッチファイルの初期化
- L101: タイトルと分子指定セクションを読み込み。
- L102: Fletcher-Powell 最適化
- L103: 極小値やTS、STQN遷移状態探索のためのBerny 最適化
- L105: Murtaugh-Sarent 最適化
- L106: 分極率および超分極率を得るための力および双極子の数値微分
- L107: LST (Linear-synchronous-transit) 法による遷移状態探索
- L108: 緩和させない (unrelaxed) ポテンシャルエネルギー曲面スキャン
- L109: Newton Raphson 最適化
- L110: 振動数を得るためのエネルギーに対する二次数値微分
- L111: 分極率および超分極率を計算するためのエネルギーに対する二度の数値微分
- L112: 自己無撞着なビリアルスケーリング法 (Self-Consistent Virial Scaling method, SCVC) の実行 (T. A. Keithの表式[Lowdin59, Magnoli82, Lehd91])
- L113: 解析的勾配を用いたEFアルゴリズムによる構造最適化
- L114: EFアルゴリズムによる数値的な構造最適化 (エネルギーのみを利用)
- L115: GS3アルゴリズムによる反応経路探索
- L116: 数値的な自己無撞着反応場 (self-consistent reaction field、SCRF)
- L117: IPCM溶媒和計算の実行
- L118: BOMD計算
- L120: ONIOM計算の操作
- L121: ADMP計算
- L122: Counterpoise計算
- L123: HPCアルゴリズム (やその他) を用いた反応経路探索
- L124: PCMおよびexternal-iteration PCMを用いたONIOM計算の実行
- L202: 座標系の再設定、対称性の計算、変数の確認
- L301: 基底関数情報の生成
- L302: 重なり積分、運動および位置の積分の計算
- L303: 多極子積分の計算
- L308: 双極子の速度とRx∇積分の計算
- L310: 原始型spdf タイプの2電子積分計算
- L311: spタイプの2電子積分計算
- L314: spdfタイプの2電子積分計算
- L316: 2電子積分の出力
- L319: 近似のスピン軌道カップリングについての1電子積分の計算
- L401: 初期MO係数 (guess) の生成
- L402: 半経験的 (Semi-empirical) 分子軌道計算や分子力学 (Molecular Mechanics) 計算の実行
- L405: MCSCF計算の初期化
- L502: SCF方程式の繰り返し計算 (conventional UHF、ROHF、すべてのDirect法、SCRF)
- L503: ダイレクト最小化を用いたSCF方程式の繰り返し計算
- L506: ROHFやGVB-PP計算の実行
- L508: 二次的な収束SCFプログラム
- L510: MC-SCF
- L601: ポピュレーション解析および関連する解析 (多極子モーメントを含む)
- L602: 1電子プロパティ (ポテンシャル、電場、電場の勾配)
- L604: グリッド点上のMOや電子密度の見積もり
- L607: NBO解析の実行
- L608: 繰り返しでないDFTエネルギー
- L609: Atoms in Moleculesプロパティ
- L610: 数値積分 (積分コードのテスト)
- L701: 1電子積分の一次または二次微分
- L702: 2電子積分の一次または二次微分 (sp)
- L703: 2電子積分の一次または二次微分 (spdf)
- L716: 構造最適化と振動数についての情報を処理
- L801: 2電子積分変換の初期化
- L802: 積分変換の実行 (N3 in-coreアルゴリズム)
- L804: 積分変換
- L811: 積分導関数の形成、MP2の二次微分の寄与の計算
- L901: 2電子積分の反対称化
- L902: Hatree-Fock波動関数の信頼性の判定
- L903: (旧) in-core MP2
- L904: Peterssonらによる完全基底関数 (CBS) 外挿法
- L905: 複合的なMP2
- L906: セミダイレクトMP2
- L908: 電子伝播 (Electron Propagator) プログラム
- L909: ADC(3) と関連する電子伝播モデル
- L913: ポストSCFエネルギーとその勾配項の計算
- L914: CIS、RPA、ZIndo励起状態とSCF安定性
- L915: 五次のオーダーの手法 (MP5、QCISD (TQ) 、BD(TQ)) の計算
- L916: (旧) MP4とCCSD
- L918: 波動関数の最適化の再計算
- L923: SAC-CI プログラム
- L925: 励起状態の電子移動 (EET) モデルの実行
- L1002: CPHF方程式の繰り返し計算とNMRを含む様々なプロパティの計算
- L1003: CP-MCSCF方程式の繰り返し計算
- L1101: 1電子積分の導関数計算
- L1102: 双極子モーメントの積分および微分の計算
- L1110: F(x)に対する2電子積分の微分の寄与
- L1111: 2粒子の密度行列およびポストSCFの微分
- L1112: MP2の2次微分
- L9999: 計算と出力の終了
新しい機能
バージョン履歴
製品概要
追加された機能
動作環境
Gaussian 16 UNIX/Linux/macOS バージョン
最新の動作環境は下記 Gaussian 社サイトをご覧ください。
Gaussian 16 Windows バージョン
Gaussian 16W は、Gaussian 16 の Windows 版です。Gaussian 16 そのものが Windows 環境に実装されています。Gaussian 16W は、32-bit 版と 64-bit 版をご選択いただけます。詳しくはライセンスをご覧ください。
推奨されるシステムの最小構成
32-bit 版
Gaussian 16W 32-bit 版は、システムに搭載されたメモリ容量にかかわらず、利用できる RAM は最大 2 GB、ディスクは最大 16 GB となります。32-bit マルチプロセッサ版が利用可能なプロセッサ数 (コア数) は 4 つが上限となります。
- OS: Windows 7 / 8 / 8.1 / 10 / 11, Windows Server 2012 R2
※ 64-bit 環境では 32-bit モードで動作します。 - CPU: Intel Pentium 4, AMD Athlon 以上
- メモリ (RAM) : 1GB
- ディスク容量 : G16W 用に 1.7GB、スクラッチ空間として更に 500MB 以上
- その他 : DVDドライブ、マウス
64-bit 版
Gaussian 16W 64-bit 版は、SMP 計算において利用できるプロセッサ数 (コア数) には制限はありません。
- OS: Windows 7 / 8 / 8.1 / 10 / 11, Windows Server 2012 R2
- CPU: AMD64 または Intel64 (EM64T) 以上
- メモリ (RAM) : 2GB 以上
- ディスク容量 : G16W 用に 1.5GB、スクラッチ空間として更に 2GB 以上
- その他 : DVDドライブ、マウス
Gaussian 16 macOS バージョン
※ Gaussian 16M は、ターミナルアプリケーションです。インストールおよび実行には、UNIX コマンドの知識が必要です。
Apple macOS システムで Gaussian 16 を利用するには、次の 2 つのバージョンの中のいずれかをお選びいただけます:
- Gaussian 16 UNIX
通常の Gaussian 16 の UNIX バージョンは、64-bit 演算およびマルチプロセッサ/マルチコア・システムを含む macOS システムをサポートします。ライセンスの締結は、Linux/UNIX と同じ手続きとなります (詳しくはこちらをご覧ください。) 。ネットワーク/クラスタ並列バージョン (Linda 並列バージョン) もご利用いただけます。動作環境は Linux/UNIX の情報を確認してください。 - Gaussian 16M シリアル版
Gaussian 16M シリアル版は、macOS システムで利用できるシングル CPU バージョンの Gaussian 16 です。システムに搭載されたメモリ容量にかかわらず、利用できる RAM は最大 2 GB、ディスクは最大 16 GB となります。シュリンクラップ・ライセンス (開封により使用許諾に同意したものとみなされるライセンス) 製品として提供されます。
アプリケーション配信、シンクライアント、ネットブート、仮想環境のサポートについて
- 弊社取り扱い製品すべてを対象に、アプリケーション配信、シンクライアント、ネットブート、仮想環境における動作は正式サポートしておりません。
これらのシステムの運用に関連する致命的な問題は報告されておりませんが、問題が生じたとしても対応致しかねる場合がありますことをご了承下さい。 - 上記運用に関してご不明な点がありましたら、弊社営業部 ( soft.sales@hulinks.co.jp ) 迄お問い合わせ下さい。
ライセンス
Gaussian 16 は、使用するコンピュータの OS によってライセンスの種類が分かれます。詳細については、それぞれのライセンスの説明をご覧ください。
UNIX/Linux 版(Gaussian 16)
- UNIX、Linux、Intel Mac コンピュータで使用する製品です。
- 共有メモリ・マルチプロセッサシステム環境における並列実行も可能です。
- クラスタおよびネットワークを介して並列実行する場合は、並列演算環境ソフトウェア Linda のライセンスが別途必要となります。
- サイトライセンスが基本ですが、一般用にはシングルマシンライセンスもございます。
- 購入に際しては開発元とお客様の間で契約(英文)の締結が必要です。
詳細はこちらをご覧ください。
Windows 版(Gaussian 16W)
- Windows コンピュータで使用する製品です。
- 32-bit 版と 64-bit 版がありますが、両者はライセンス体系が異なります。
32-bit 版
- 32-bit 版のシングルマシン版は、シュリンクラップ・ライセンス(開封により使用許諾に同意したものとみなされるライセンス)です。契約(英文)の締結は必要ありません。
- シングルプロセッサまたはコアで Gaussian 16W を実行できるシリアル版と、マルチプロセッサまたはマルチコア(共有メモリ)システムを使ったプログラムの並列実行が可能なマルチプロセッサ版の2種類が用意されています。
- マルチプロセッサ版にはサイトライセンスもあります。
64-bit 版
- 64-bit 版には、シュリンクラップ・ライセンスはありません。購入に際しては開発元とお客様の間で契約(英文)の締結が必要です。
Gaussian 16W のサイトライセンス
Gaussian 09W Revision B.01 以前に購入
- 32-bit 版/マルチプロセッサ版のサイトライセンスです。
- 64-bit 版を含むサイトライセンスへのアップグレードが可能です。
Gaussian 09W Revision C.01 以降に購入
- 32-bit 版、64-bit 版の両方が使用できるサイトライセンスになりました。
Intel Mac 版(Gaussian 16M)
- 32-bit 版のシリアル版に限定した Intel ベースの Mac OS X コンピュータ用の製品です。
- シュリンクラップ・ライセンスですので、契約(英文)の締結は必要ありません。
- 64-bit 版や、共有メモリ・マルチプロセッサシステム環境における並列実行を希望される場合は、UNIX/Linux 版の Gaussian 16 をご購入ください。
マイナーリビジョンについて
- マイナーリビジョンとは
Gaussian には、メジャーバージョン(現在は 16)に加え、マイナーリビジョンと呼ばれるマイナーバージョンが設定されています。
例;Gaussian 16 Revision: C.02
おもに対応環境の変更やその他の変更が行われた場合に、マイナーリビジョンのバージョンアップが行われます。 - マイナーリビジョンのアップグレード方法
Gaussian 年間メンテナンスにご加入されているユーザー様には、Gaussian 社からマイナーリビジョンのメディアが無償で送付されます。
メンテナンスにご加入されていないユーザー様がマイナーリビジョンのアップグレードをご希望される場合は、新しいマイナーリビジョンのメディアをご購入していただく必要があります (Minor revision media fee) 。
なお、シングルライセンスのマイナーリビジョンのメディアは、インストール時に過去のバージョンがインストールされていることが必要になりますのでご注意ください。
詳しくは、弊社営業部 (soft.sales@hulinks.co.jp) までお問い合わせください。
各ライセンスの詳細
製品価格
ご購入時における注意点
お申込みはご所属の組織経由で
- Gaussian 製品は、シングルマシンライセンスであってもご所属の組織 (企業・大学・研究所など) 経由でのご注文が基本となります。(個人でのご注文は Gaussian 社に事前確認が必要です。)
契約書の締結について
Gaussian 製品には、シュリンクラップライセンス(開封と同時に使用許諾に同意したものとみなされるライセンス。契約書不要)と契約書の締結が必要なライセンスがあります。
- シュリンクラップライセンス
- Gaussian 16W 32-bit シリアル版 シングルマシン(一般用/教育用)
- Gaussian 16W 32-bit マルチプロセッサ版 シングルマシン(一般用/教育用)
- Gaussian 16M/IntelMac シングルマシン(一般用/教育用)
- 上記製品と GaussView 6 の Bundle 製品
上記以外のすべての Gaussian 製品は、ご購入に際し、Gaussian 社との間で契約書をご締結いただきます。なお、契約書には「サイト (同じ住所にある同一組織) を代表される方」にサインしていただく必要があります。
「サイトを代表される方」の定義
- Gaussian 社製品の契約書のサインは、ご担当者様ではなく、サイトの管理権限のある方(サイトを代表して契約を締結する権限を有する方)にお願いいたします。 Gaussian 社が認めている肩書きは次のとおりです。実際の職務権限から判断していただき、該当する方に次のいずれかの英文肩書きを使用してサインをお願いいたします。
| Gaussian 社が認める英文肩書き | 認められない英文肩書き |
| President、Vice-President、Treasurer (Other) Corporate/Institutional Officers Purchasing Agent(not departmental) Rector Provost Dean of Faculty/College | Department Chair Professor Researcher Lecturer Director ※ |
製品の発送について
- 製品は Gaussian 社からエンドユーザー様に直送されます。購入申込書、ユーザー情報確認書でエンドユーザー様情報 (日本語・英語) をお知らせ願います。
既にサイトライセンスをお持ちの場合
お客様が Gaussian 製品をご注文されると、所属されるサイトがご希望される製品のサイトライセンスを所有していないか Gaussian 社で確認が行われます。該当するサイトライセンスがない場合は、そのままご購入の手続きが進みます。一方、該当するサイトライセンスをお持ちであることが分かった場合には、次の中からお客様のご希望を確認させていただいた上で手配を進めます。
- シングルマシンライセンスをご注文いただいていた場合:
以下の選択肢があります。- 既存のサイトライセンスは使用せず、シングルマシン版を購入する。(DVDが付きます。)
- 既存のサイトライセンスを使用する。(※1 )
※1 :既存のサイトライセンスを使用する場合のプログラムの入手方法
- サイトライセンスの管理者からメディアを借りる。
→ ご購入いただくものはございませんので、ご注文はキャンセルとなります。 - 最新のメディア(DVD)を購入する。
→ ご注文内容は、Minor Revision Media Fee に変更となります。
※ サイトライセンスの範囲内でメディアをご購入されたお客様には、弊社の無償テクニカルサポートサービスはご提供いたしかねます。ご了承ください。
お見積り・ご購入
サポート
Gaussian 社 Annual Maintenance (年間メンテナンス)
Gaussian 社では、原則としてサイトライセンスを対象とした Annual Maintenance (年間メンテナンス) プログラムを用意しています。
このプログラムは、プラットフォーム毎ではなく、ご購入いただいた全ての Gaussianサイトライセンスをカバーします。
メンテナンスご加入のメリット
- Gaussian 社のスタッフによるテクニカルサポート(英語)を優先的に受けることができます。
- メンテナンス期間内にマイナーリビジョンおよびメジャーバージョンのアップグレードがあると、 Gaussian 社からメディアが無償で送付されます。
- メジャーバージョンのベータテストに参加することができます。
詳しくは、Gaussian, Inc. Maintenance Program または、弊社営業部 (soft.sales@hulinks.co.jp) までお問い合わせください。
ヒューリンクス・日本語サポートサービス
ヒューリンクスでは下記のサービスを提供しています。お問い合わせは弊社営業部 (soft.sales@hulinks.co.jp) まで。
無償サポート
| サポート | 対象製品 | 期間 | 費用 |
|---|---|---|---|
| 無償サポート | G16 W / M シングルマシン GV6 W / M シングルマシン | – | |
サポート範囲
| |||
ご注意
| |||
Gaussian 日本語技術サポート (有償サポート)
| サポート | 対象製品 (下記より一製品ご指定下さい) | 期間 | 税込費用 |
|---|---|---|---|
| Gaussian 有償サポート | |||
| G16 (UNIX/Linux, Windows, Mac) サイトライセンス | 1年 | ¥330,000 | |
| G16 (UNIX/Linux, Windows, Mac) シングルマシン | 1年 | ¥132,000 | |
| G16 (UNIX/Linux, Windows, Mac) シングルマシン | 半年 | ¥66,000 | |
サポート範囲
| |||
ご注意
| |||
Gaussian + GaussView アドオン 日本語技術サポート (有償サポート)
| サポート | 対象製品 (下記より一製品ご指定下さい) | 期間 | 税込費用 |
|---|---|---|---|
| Gaussian + GaussView 有償サポート | G16+GV6 (UNIX/Linux, Windows, Mac) サイトライセンス | 1年 | ¥442,200 |
| G16+GV6 (UNIX/Linux, Windows, Mac) シングルマシン | 1年 | ¥198,000 | |
| G16+GV6 (UNIX/Linux, Windows, Mac) シングルマシン | 半年 | ¥99,000 | |
| オプション | GMMX アドオン (上記サイトライセンス用) | 1年 | ¥132,000 |
| GMMX アドオン (上記シングルマシン・1 年用) | 1年 | ¥66,000 | |
| GMMX アドオン (上記シングルマシン・半年用) | 半年 | ¥33,000 | |
サポート範囲
| |||
ご注意
| |||
インストール・サービス
| インストール・サービス | 対象製品 | 費用 |
|---|---|---|
| 出張インストール | Gaussian ソースコード | お問い合わせください。 |
| Gaussian バイナリ (UNIX/Linux, Windows, Mac), GaussView | お問い合わせください。 |
※ 最終的な費用決定はお客様の状況確認後となります。
※ Linda も必要な場合はご相談ください。
最適な計算環境の構築の支援サービス
ヒューリンクスでは、Gaussian/GaussView を実行するためのハードウェアも取り扱っております。詳しくは弊社 (soft.sales@hulinks.co.jp) までお問い合わせください。
インストールについて
- Gaussian 16 W インストールガイド
- Gaussian 16 UNIX Binary インストールガイド
- Gaussian 16 macOS Binary インストールガイド
- GaussView W 6 インストールガイド
- GaussView 6 for UNIX インストールガイド
- GaussView 6 for macOS インストールガイド
- GMMX アドオン インストールガイド (Windows 版)
旧バージョン
パフォーマンスについて
その他
- Gaussian 16 と GaussView 5 、Gaussian 09 と GaussView 6 の組み合わせでの利用について
- Gaussian ユーティリティについて
- バッチ処理中のエラー中断を無効にして計算を続行させる方法
- 振動計算結果を GaussView に読込む際に表示されるメッセージについて
- Severe Error #2070 について