|
| サイトマップ | |
||
|
| サイトマップ | |
||
Mac 版および Windows 版 CrystalMaker 9.1 の機能が大幅に向上しました。本ドキュメントでは新機能について説明し、既存のユーザーズガイドを補足します。
CrystalMaker 9.1 は、新しいファイルフォーマット、モデリングの改善、より柔軟な範囲設定と結晶工学ツールを備え、すべての領域でプログラムの機能が向上しています。また、新しいキーボードショートカット、新しいプロット範囲レポートやバグフィックス等、多くのマイナーチェンジも含んでいます。
CrystalMaker 9.1 の新機能によって、結合鎖の構築、水素原子の追加、大きな分子構造で選択した原子だけを緩和することがより簡単になりました。
 |
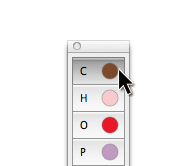
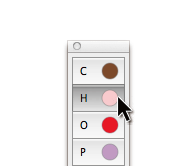
Add Atom ツールを使って、原子と結合による分子の主鎖を定義することができるようになりました。shift キーを押しながらクリックするだけで、新しい原子とそれより前に追加した原子の間に結合が追加されます。これによって、結合鎖を素早く構築することができるだけでなく、追加の原子または側鎖を簡単につなぐことができます。
Add Atom ツールを使ってクリックすると、各々の新しい原子がハイライト表示され、新しい結合の「アンカー」であることが示されます(シフトクリックすることによって新しい原子が追加されます)。Add Atom ツールで、別の原子をクリックすると、「アンカー」をリセットすることができます。たとえば、側鎖をポリマー分子に加えるためには、ポリマー中の1つの炭素原子をクリックし、次に、シフトキー+クリックで新しい炭素原子とその結合を加えることができます。もう一度シフト+クリックをすると、側鎖を長くすることができます。
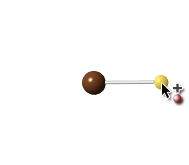
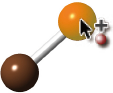
Add Atomツールを使って(緩和されていない)メタン分子を作成
|
|
|
|||
|
|
|
|||
|
|
|
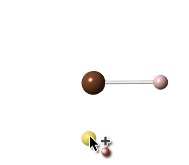
さらに分子を広げる場合は、炭素原子の間でシフト+クリックして主鎖を定義し、Add Hydrogens コマンドを使用します(詳細は次のセクションをご参照ください)。
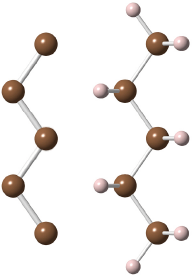 |
CrystalMaker の新しい Transform > Add Hydrogens コマンドを使用して、水素原子をどのようなな分子にも自動的に追加できるようになりました。プログラムが、構造が完全飽和する必要があると仮定して、新しい水素の上で緩和を実行し、最適なジオメトリを生成します(既存の原子の位置は緩和されません。そのため、このコマンドはX線データを使用した精密な分子構造に水素原子を追加するのに適しています)。
水素の位置は素早く精緻化しなければなりませんが、緩和を停止したい場合は、ウインドウのツールバーで Stop Relaxing ボタンをクリックするか、または Transform メニューから Stop Relaxing コマンドを選んで停止させることができます。
| Tip:二重または三重結合にしたい(例えば、C―O ユニットに水素を加えて H3C―OH の代わりに H2C=O を作成する)場合、新しく追加した水素原子のいくつかを(隠すのではなく)削除してください。原子を削除するには、削除する原子を選んで、delete キーを押してください(または Selection > Delete を使用してください)。 |
構造全体ではなく、構造の一部(例えば、分子の側鎖または表面の原子)を緩和させることが可能になりました。緩和させたい原子を選んで、ウィンドウのツールバーで Relax ボタンをクリック(あるいは、Transform > Relax Selection を選択)するだけです。
CrystalMaker 9.1 では、結晶内の様々な範囲の原子を調査し、最初の選択に基づいて、個々の座標環境または全ての分子を修復して、非対称ユニットを単離させ、すべての(目に見える)左右対称の原子を簡単に選択できるようになりました。
プロット範囲を変えても、原子の属性が保持されるため、カスタムの原子の色、スタイル、ラベルなどが保持されます。(CrystalMaker のこれまでのバージョンでは、結晶格子の新しいブロックを生成する必要があると、カスタムの原子がリセットされていました。)
| 正のフラクショナル座標系 |
| CrystalMaker は常に正のフラクショナル座標系を用います。0 番目のユニットセルから始まり、正の x、y、z 結晶軸に沿って増加します。負の座標を Set Range ウィンドウに入力した場合、構造がプロットされると正の値に変換されます。 |
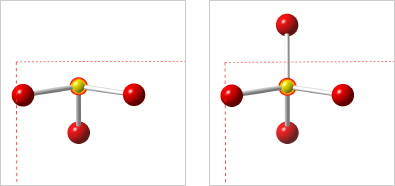
不完全な結合を持つ原子は配位を修正することができるようになり、最近接原子とその結合を可視化できます。同様に、「壊れた」分子を修復できます。どちらの場合も、原子と結合の欠落はプロット範囲の切断によって生じます。CrystalMaker は、適切な方向にプロット範囲をインテリジェントに拡大して選択した原子を「育て」、残りの構造を同じ状態に保ちます。
原子の周辺の最近接原子結合の修復方法:
 |
「壊れた」分子の修復方法:
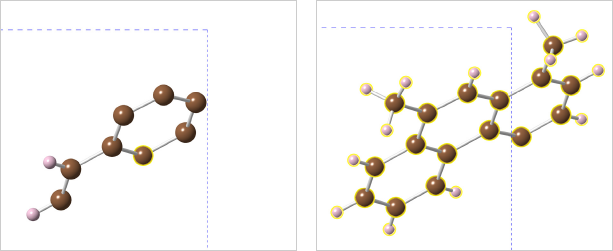 |
CrystalMaker を使って、より簡単に分子結晶の中の一つの分子を単離化することができるようになりました。新しいメニューコマンド(Transform > Optimize Range > Show Asymmetric Unit)を使うと、オリジナルの非対称ユニットが(未加工のテキストファイルからロードしたとおりに)表示されます。ほとんどの分子結晶の場合、これは一つ以上の無傷の分子に対応します。
| Tip:この新しいコマンドは既存の Show Molecular Cell コマンドを補完し、1つのユニットセルの完全な内容を表示し、すべての分子が無傷であることを確実とするために最適化されます。 |
注意:非対称ユニットのフラクショナル座標系が変更された場合(CrystalMaker の初期バージョンで、自動的にサイトを同じユニットセルにまとめた場合等)、結果が予測できない可能性があり、分子が壊れて表示される可能性があります。
 |
結晶の中の対称に関係するすべての原子を素早く選択することができます。まず1つの原子を選んで、新しいメニューコマンド:Selection > Select > Symmetry-Related Atoms を選んでください。すべての対称と等価の原子(つまり、非対称ユニットにある同じサイトを持つ原子)が選択されます。
| 結合に関連する対称 |
| 原子の組を選び、Select Symmetry-Related Atoms コマンドを使うと、対称に関連する結合をハイライト表示することができます。たとえば、2種類の Si 原子、T1 と T2、4種類の酸素原子 O1…4 を持つ構造がある場合を考えます。1つの T1–O1 結合を選択し、CrystalMaker を使ってすべての等価な組を選ぶことができます。これによって、現在のプロット範囲内のこれら特定の結合を強調するために、カスタムの結合のスタイルや色を指定することができます。 |
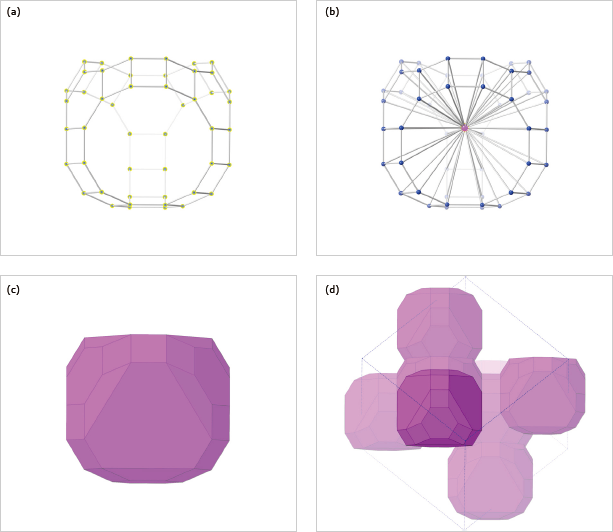
Make Polyhedronコマンドが、新しいデフォルトの結合オプション ”All bonds in this distance range” で改良されました。これによって適切なグローバル結合仕様が、選択した距離範囲に基づいて生成され、その結果、プロット範囲が変更されても多面体は保持されます。
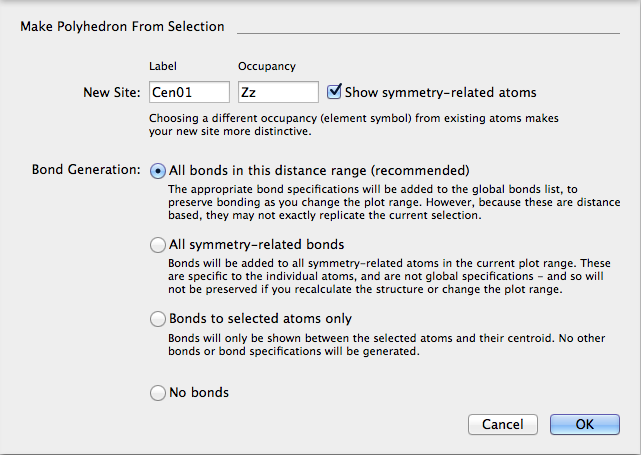 |
たとえば、アルミノケイ酸塩の「ケージ」を「純粋な多面体」を使用したゼオライトで表したい場合は、Al/Si ケージ原子を選択して、Make Polyhedron コマンドを選ぶだけです。新しいデフォルト・オプションが、ケージ原子からケージ重心までの結合距離の範囲を分析し、プロット範囲内のこの多面体を伝播するグローバルな結合の仕様を生成します。
新しいデフォルトの手法は、ほぼ球状の座標環境のほとんどの場合に機能します。しかし、形をさらに変形させている場合は、意図しない結合が作成される以上の危険があります。このような場合は、All symmetry-related bonds オプションを選択することをお勧めします。ただ、これは、現在のプロット範囲にのみ適しています。プロット範囲を広げて、結晶格子を再作成させた場合はリセットされる可能性があります。
 |
 |
新しい結晶工学ツールの一部として、“crystal” モードで選択した原子グループを回転させることができるようになりました(以前は “molecule” モード限定の機能でした)。選択を回転させるためには、Tools パレットの右下隅のボタンを使用して Rotate Selection モードに切り替えてください。その後、Rotate ツールまたは Rotator Pad を使用して、構造の残りの部分に対応して選択部分を回転することができます。
なお、格子のタイル化の理由で、選択部分が1つのユニットセルを越えている場合は、回転ができないことがあります。
CrystalMaker は、ポピュラーなエネルギーモデリングプログラムである LAMMPS および GROMACS で作成された結晶構造ファイルをインポートできるようになりました。これらのフォーマットは自動判別されるため、データファイルをプログラムにドラッグ&ドロップして即座に表示することができます。他のファイル形式と同様に、CrystalMaker はマルチ構造ファイルをサポートしているため、Overview ウィンドウの Views 枠にサムネイルとして個々の構造を表示します。
LAMMPS は、米国のサンディア国立研究所で開発されたエネルギーモデリングプログラムです。CrystalMaker は、以下のような座標 “dump” フォーマットに対応しています:
ITEM: TIMESTEP 11000 ITEM: NUMBER OF ATOMS 8000 ITEM: BOX BOUNDS pp pp pp 0 18.8207 0 18.8207 0 37.6414 ITEM: ATOMS id type xs ys zs 7936 1 0.0515603 0.00797728 0.00467763 6719 1 0.00242674 0.0395856 0.0162416 7837 1 0.1169 0.0539664 0.00190204
注意:LAMMPS は要素シンボルを出力ファイルに埋め込むことはできません。数値である原子 ID だけです。CrystalMaker は、完全に任意のシンボルを割り当てます。Edit Crystal ウィンドウを使用するか、Selection > Atoms > Change Atom Type で変更したい原子を選択して変更することができます。
GROMACS は、生体システム用に設計されたエネルギーモデリングプログラムです。サンプルファイルは以下のようになります:
MD of 2 waters, t= 0.0 6 1WATER OW1 1 0.126 1.624 1.679 0.1227 -0.0580 0.0434 1WATER HW2 2 0.190 1.661 1.747 0.8085 0.3191 -0.7791 1WATER HW3 3 0.177 1.568 1.613 -0.9045 -2.6469 1.3180 2WATER OW1 4 1.275 0.053 0.622 0.2519 0.3140 -0.1734 2WATER HW2 5 1.337 0.002 0.680 -1.0641 -1.1349 0.0257 2WATER HW3 6 1.326 0.120 0.568 1.9427 -0.8216 -0.0244 1.82060 1.82060 1.82060
注意:GROMACS は要素シンボルをユニークには定めず、原子ラベルに依存します。CrystalMaker では、構造が有機的であると仮定して、原子ラベルの最初の文字を使って、要素シンボルを割り当てます(例えば、ラベル「HE1」を持つ原子は「He」ではなく「H」と解釈されます)。