|
| サイトマップ | |
||
|
| サイトマップ | |
||
このルーチーンは、2つのシーケンスの配列全体を使用してそれらを最適に整列 (alignment) するものです (大域的アラインメント)。整列パラメータは、一致、ミスマッチ (不一致)、ギャップの挿入、ギャップの伸長について定義することができます。このルーチーンは、2つのグロブリンのように関連性の高いことが既知である2つのシーケンスを整列する場合に最適です。"末端ギャップ (end gaps)" に対するペナルティはオプションにより選択可能で、また、Z-score を計算して整列結果の有意性を調べることもできます。Z-score、末端ギャップ、およびその他のパラメータに関する詳しい説明については、核酸セクション の “Align 2 Sequences (Global)” をご覧ください。
タンパク質における大域的整列 (global alignment) と核酸におけるそれとの違いの一つは、タンパク質の大域的整列の方には、進化モデルに基づいて綿密に定義されたスコアテーブルがいくつか存在する点にあります。PAM および BLOSUM という名称の付いたテーブルがそれです。
PAM テーブルは、Dayhoff らの論文 [in “Atlas of Protein Sequence and Structure”, vol 5 supplement 3, ed. M.O. Dayhoff, NBRF, Washington D.C., (1978), p345] のデータを元に作成されたものです。これは、近縁の多数のタンパク質のセットを対象に、あるアミノ酸が進化の過程で別のアミノ酸に変異する頻度を調べたものです。これらの近縁のタンパク質で観察された変異の数に基づいて、タンパク質配列の同じ部位で連続的変化が生じると考えられることから、それよりも進化的に遠い距離で観察される変異数を見積もる方法が開発されました。PAM1 テーブルは、タンパク質内でアミノ酸 100 につき1つの変異があるとして、あるアミノ酸が別のアミノ酸に変換する確率をまとめたものです。この PAM1 テーブルは確率をあらわしたテーブルなので、PAM 1 行列をそれ自身と 40 回掛け合わせることで、アミノ酸 100 につき 40 の点突然変異を許容する比較表 PAM40 行列ができあがります。
| Evolutionary Distance in PAMs | Observed Percent Difference |
|---|---|
| 1 | 1% |
| 5 | 5% |
| 11 | 10% |
| 17 | 15% |
| 23 | 20% |
| 38 | 30% |
| 56 | 40% |
| 80 | 50% |
| 112 | 60% |
| 159 | 70% |
| 246 | 80% |
| ※ *. This table is from “Molecular Sequence Comparison and Alignment” by J.F. Collins and A.F.W. Coulson in “Nucleic Acid and Protein Sequence Analysis: A Practical Approach”, ed M.J. Bishop and C.F. Rawlings, IRL Press, Washington, D.C.(1987), p323. |
Gene Inspector では PAM テーブルとして、PAM40、PAM120 および PAM250 があらかじめ用意されています。アミノ酸 100 につきシーケンス内で変異が 250 起こるほど遠い進化的距離にあるシーケンスでも実際に認識することができるのです!シーケンス同士が近縁であれば、PAM の値の小さいテーブルを使用して比較しますが、進化的に距離の離れたシーケンスを比較するには PAM 値の大きいテーブルを使用してください。上に示したテーブルに表示されているとおり、突然変異の発生数を多く割り当てても、それらは同じ位置で繰り返し発生しますので、PAM246 (100 残基あたりの突然変異が 246) でもシーケンス内で変異するアミノ酸の割合は 80% にとどまります。残りの 20% でシーケンスの類似度を十分認識することができます。
もうひとつのテーブルセットは、[S. Henikoff & J.G. Henikoff, Proc. Nat. Acad. Sci. USA 89:10915 (1992)] に依拠した BLOSUM テーブルです。BLOSUM テーブルは、関連のあることが既知の 500 を超えるタンパク質群を整列してできたブロックと呼ばれるシーケンスセグメントを元に作成されたものです。BLOSUM62 がシーケンスを比較する最適な手段のひとつを提供し、PAM 行列を使用して比較するより優れた結果になることは、 [Henikoff & Henikoff, Proteins 17:49 (1993)] で実験されています。近縁のタンパク質の既知の類似性に基づいて、進化的に距離の離れたタンパク質の類似性を予測するという PAM 行列が開発された考え方より、BLOSUM テーブルの方が高い精度であることが分かっています。一般に、BLOSUM62 行列を使って比較を始めるのが最適であるとされています。PAM テーブルを使って行うのと同様、シーケンスどうしが近縁であるほど、小さい数値の BLOSUM テーブルを使うようにしてください。進化的に距離の離れたシーケンスを比較する場合は、数値の大きい BLOSUM テーブルを使用してください。
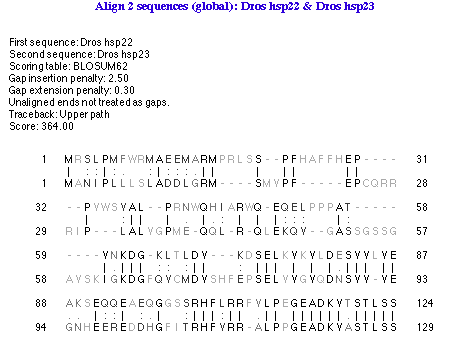
図 4.48 に示すのは、この整列による出力結果です。この事例では、類似性の最も高い領域が整列インジケーターで示されています。
 |