|
| サイトマップ | |
||
|
| サイトマップ | |
||
このルーチーンは、2つのシーケンスの配列全体を使用してそれらを最適に整列 (alignment) するものです (大域的アラインメント)。この解析により可能な限り高い「スコア (評価点)」を含む整列が作成されます。スコアは一致した各ヌクレオチドの点数から、ギャップとミスマッチの点数を差し引いて計算されます。これらの値は、図 4.12 に示すパネルで定義することができます。このセットアップパネルは多数の部分に分かれています。
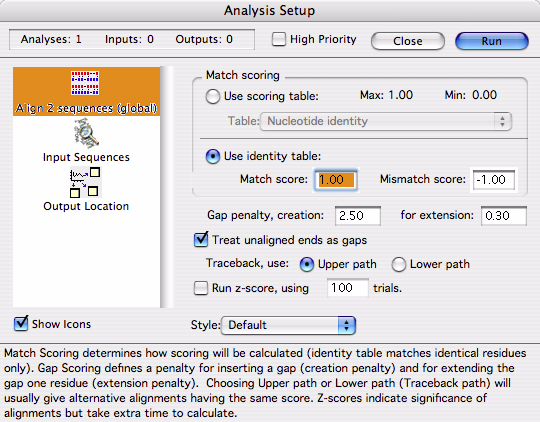 |
このパネルの一番上の部分では、実行するスコアリングの方法を指定します。スコアリングに使用するテーブルには、前回定義したスコアリング・テーブル、または、同一テーブル (identity table) を選択できます。Gene Inspector には標準的なスコアリング・テーブルがあらかじめ多数用意されていますが、Gene Inspector の Table Editor を使用すれば、スコアリング・テーブルまたは独自のテーブルを作成することもできます (詳細は、オリジナル解析テーブルの作成、および、“Tables”, page 6-23 をご覧ください)。スコアリング・テーブルには、整列スコアに加算する値を定義して、その値を各比較に使います。例えば、一致するものにはすべてスコア1を定義し、プリンとプリンの一致、または、ピリミジンとピリミジンの一致にはスコア 0.5 を付けるという指定もあるでしょう。同一テーブル (identity table) を使えば、完全一致に加算する値と、ミスマッチで差し引く値をそれぞれ指定することができます (図のようにミスマッチのスコアとして負の値を入力するだけです)。
ギャップのスコアリングでは、ギャップの挿入 (creation) と伸長 (extention) に対してどのようにスコアを差し引くかを定義することができます。ギャップ挿入ペナルティは、シーケンスのいずれか一方にギャップがヌクレオチド (塩基) 一つ分作成される度にスコアから差し引く値です。ギャップ伸長ペナルティは、ギャップ内にあるヌクレオチド (塩基) 数をスコアから差し引く値です。生物学的背景によって、ギャップ伸長ペナルティをゼロにすることが良いこともあれば (個々のヌクレオチドの連続的な挿入や欠失ではなく、単発的に生ずる DNA セグメントの挿入や欠失である場合)、ゼロ以外の値にするのが良い場合もあります。
2つのシーケンスの全長、すなわち、各 DNA の始まりと終わりの両端を整列したい場合は、Treat unaligned ends as gaps を選択します。このチェックボックスにチェックを入れると、一方のシーケンスがもう一方のシーケンスより前に開始 (または終了) している場合は、ギャップペナルティが整列スコアから差し引かれます。別の言葉で言い換えると、整列されていない末端セグメントは、ギャップとして取り扱われることになります。このオプションにチェックが入っていなければ、両端の整列処理は行われません。
同一のスコアの整列が複数作成されるのは良く起こることです。異なるトレースバックパス (Upper または Lower トレースバックパス) を選択すれば、同じ最大スコアを出力する最も異なる整列結果を確認することができます。トレースバックパス (Traceback path) とは、プログラムが生成する実際の整列作成法に相当する専門用語です。
最後に用意されたオプションは Z-score の計算です。これは、整列スコアの有意性を統計的にあらわすものですが、その処理には時間が掛かります。2つのシーケンスの整列を行う際、選択したスコアリング・テーブルと解析によって生成された整列結果に基づいてスコアが計算されます。スコアは出力オブジェクトに表示されます。ただし、整列の有意性をこのスコアから判断するのは難しい場合もしばしばあります (247.4 というスコアが意味する内容とは?)。Gene Inspector では、こうした問題に取り組み、整列に関する何らかの統計情報を提供するために Z-score を計算するための手段が用意されています。
Z-score の計算は、幾つかのステップを経て行います。まずはじめに、シーケンスの一方 (これをシーケンス A と呼びましょう) を「シャッフル」します。つまり、そのシーケンスを構成する塩基の並びをランダムにごちゃ混ぜにします。これにより、元の塩基配列は失われますが塩基組成 (base composition) は保持されます。ここで、シャッフルしたシーケンス A とシャッフルされていないシーケンス B を使って整列を実行し、その整列スコアを計算します。同じプロセスをシャッフルされた別のバージョンの A とシャッフルしない B についても繰り返します。このプロセスを何回も繰り返し、整列スコアの平均と標準偏差を求めます。Z-score は、真の整列スコアが、シャッフルした整列スコアの平均から標準偏差で幾つ分離れているかをあらわすものです。また、シャッフルしたシーケンスを用いた整列の方が、シャッフルしていないシーケンスを用いた真の整列よりスコアが良かった数も Gene Inspector に表示されます。Z-score を計算するかどうか、反復計算を何回にするかは、インプットパネル (図 4.12) を使って指定することができます。Z-score の計算にはプログラムで多数の整列処理が必要となるので相当の時間が掛かります。
なお、配列の長いシーケンスを選択すると整列に掛かる時間が非常に長くなる場合があります。整列計算に掛かる実行時間は、2つのシーケンスの長さの積に比例します。従って、ヌクレオチド数がそれぞれ 200 の2つのシーケンスを整列するには、ヌクレオチド数がそれぞれ 100 のシーケンスを整列する時間の4倍かかることになります。Gene Inspector のソースコードには非常に長い整列の実行について特に制限は設けられていませんが、追加の空きディスク容量と、それを待つだけの辛抱を覚悟する必要があるかもしれません。テンポラリ・データを格納できるだけのディスク容量を確保できれば、他のプログラムでは整列できないような非常に長いシーケンスの整列でも Gene Inspector で実行することができます。問題はテンポラリ・データを確保できるだけのディスク容量を用意できるかどうかです (詳細は “解析における追加ディスクスペースの使用について” 参照)。
ただし、Z-score を求めるのに時間が掛かるとしても、Gene Inspector で実行する他の解析と同様、整列処理はバックグラウンドで実行されますので、整列の計算中も他の作業を続けることができます。これに関する役立つヒントは “時間のかかる解析について” をご覧ください。
グローバル・アラインメントの出力結果は図 4.13 のようになります。
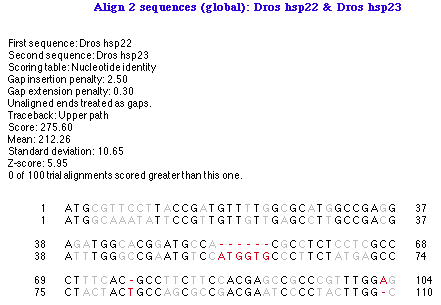 |
この解析出力オブジェクトには大きく分けて3つの部分があります。タイトル、サマリー、および、ボディ (整列結果の具体的内容) の3つです。出力オブジェクトをターゲット状態にしてあらわれる Object メニューの項目を選択することで、サマリーとボディーの表示/非表示を切り替えることができます。同じ方法を使ってg、整列インジケーターの表示と非表示を切り替えることもできます。
| ※ g. 図 4.13 には整列インジケーターは表示されていませんが、図 4.48 のペプチドの整列では表示されています。 |
整列インジケーター (Alignment indicator) とは、整列された2つの文字の関係をあらわす文字記号です。例えば、“|” はスコア 1 を表し、“:” はスコア 0.5、 “.” はスコア 0 を表すような場合です。Object > Edit Thresholds... を使えば、各スコアをあらわすカラーコードの定義はもちろん、整列インジケーターにどの文字記号を使うかを自由に定義することができます。図 4.14 に示すのはそのウィンドウです。文字記号を変更するには、変更したい “Char” ボックスをクリックして、右側のボックスに変更する文字を入力するだけです。インジケーターの色を変更するには、目的の Char を選択したあと、Format > Color を使用します。各文字記号や色に使用される閾値 (threshold) を変更するには、左側にある threshold の High, Mid, Low 各ボックスに別の値を入力します。整列インジケーターは、ペプチドの整列に大変役立ちます。
 |
{kind=link}